When discussing rare genetic disorders, Wolfram Syndrome—also known as DIDMOAD—often slips under the radar.
Its complex array of symptoms, including diabetes insipidus, diabetes mellitus, optic atrophy, and deafness, makes it a challenging condition to diagnose and treat.
But why is DIDMOAD considered Wolfram Syndrome’s hidden puzzle?
Let’s break it down.
Article Index:
- The Enigma of DIDMOAD
- What is Wolfram Syndrome?
- Decoding the Genetics: WFS1 and CISD2 Mutations
- Wolfram Syndrome Type 1 vs. Type 2: A Closer Look
- Wolfram Disease Symptoms and Diagnosis
- Living with Wolfram Diabetes: Challenges and Management
- Understanding Wolfram Syndrome Inheritance
- Current Research and Future Directions
- FAQs on DIDMOAD
- Conclusion: Solving the Puzzle of DIDMOAD
The Enigma of DIDMOAD
Imagine grappling with a mix of chronic health problems, all rooted in a single genetic defect. That’s the harsh reality for individuals with DIDMOAD, better known as Wolfram Syndrome.
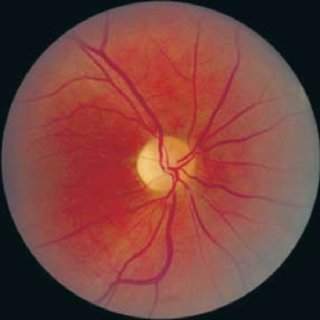
The condition is named for its primary features—Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, and Deafness—but its reach extends far beyond those four symptoms, affecting multiple organ systems.
Most commonly, Wolfram Syndrome is caused by mutations in the WFS1 gene, which disrupts cellular calcium balance and triggers endoplasmic reticulum stress, leading to widespread cell death. Symptoms often begin in early childhood.
Type 1 diabetes usually appears around age six, followed by progressive vision loss due to optic nerve damage by age eleven. Hearing impairment, difficulty with balance, and issues with urination due to a neurogenic bladder are also common.
Many individuals develop central diabetes insipidus and neurological complications such as ataxia, peripheral neuropathy, or psychiatric symptoms.
Because its symptoms overlap with other more common conditions, Wolfram Syndrome is frequently misdiagnosed, delaying access to appropriate care. With fewer than one in 500,000 people affected worldwide, it is classified as an ultra-rare disorder.
There is currently no cure, but clinical trials are exploring therapies aimed at reducing cellular stress and preserving function. These include chemical chaperones, calcium channel modulators, and even GLP-1 receptor agonists.
While treatment remains supportive, early diagnosis through genetic testing as in MODY diabetes can help manage complications more effectively. Wolfram Syndrome remains a medical mystery—rare, intricate, and in urgent need of broader awareness and research focus.
What is Wolfram Syndrome?
Wolfram Syndrome, commonly known as DIDMOAD syndrome, is a rare genetic disorder that primarily affects the endocrine and nervous systems.
The acronym DIDMOAD stands for Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, and Deafness—four hallmark symptoms of this condition.
According to the Genetic and Rare Diseases Information Center (GARD), Wolfram Syndrome is typically diagnosed in childhood or early adulthood and has a profound impact on the quality of life and life expectancy.
Decoding the Genetics: WFS1 and CISD2 Mutations
The primary genetic culprit behind Wolfram Syndrome is a mutation in the WFS1 gene, responsible for over 90% of cases.
This gene encodes the protein wolframin, which plays a critical role in maintaining cellular function by regulating calcium levels.
A malfunctioning wolframin protein leads to cellular stress and eventually cell death, particularly in insulin-producing pancreatic beta cells and optic nerve cells.
Wolfram Syndrome Type 2, a rarer form, results from mutations in the CISD2 gene, which disrupts mitochondrial function, causing symptoms like gastrointestinal bleeding and ulcers in addition to the usual features of Wolfram Syndrome.
Understanding these genetic underpinnings is crucial for developing targeted therapies in the future.

Wolfram Syndrome Type 1 vs. Type 2: A Closer Look
Wolfram Syndrome is divided into two types based on the affected genes:
- Type 1 (WFS1 Mutation): This is the more common form and includes the classic DIDMOAD symptoms. It affects insulin production and leads to early-onset diabetes, progressive vision loss, and hearing impairment.
- Type 2 (CISD2 Mutation): This type does not include diabetes insipidus but is characterized by severe gastrointestinal symptoms and a predisposition to excessive bleeding. It is primarily reported in families of Jordanian descent and remains extremely rare.
Wolfram Disease Symptoms and Diagnosis
Wolfram disease symptoms are diverse and often misinterpreted.
They include early-onset diabetes mellitus (often confused with Type 1 diabetes), progressive vision loss, hearing impairment, and neurological complications like ataxia and central apnea.
Given the wide range of symptoms, diagnosing Wolfram Syndrome can be challenging and often requires genetic testing.
Living with Wolfram Diabetes: Challenges and Management
Living with Wolfram diabetes is more complicated than managing typical diabetes due to the added burden of multiple organ dysfunctions.
Patients must regularly monitor blood glucose levels while also coping with vision and hearing loss in Wolfram Syndrome. You may even suffer urinary tract problems, and neurological complications.
The condition’s progressive nature means that management strategies must adapt over time, and there is a pressing need for multidisciplinary care.
Understanding Wolfram Syndrome Inheritance
Wolfram Syndrome follows an autosomal recessive inheritance pattern, which means a child must inherit two copies of the faulty gene—one from each parent—to develop the disorder.
If both parents are carriers of the mutated gene (usually in the WFS1 or CISD2 gene), there is a 25% chance with each pregnancy that the child will have Wolfram Syndrome, a 50% chance the child will be a carrier like the parents, and a 25% chance the child will inherit two normal copies of the gene.
Carriers typically do not show symptoms, making it possible for the condition to appear unexpectedly in families with no previous history.
Genetic testing and carrier screening become especially important in cases where there is a known family history or consanguinity, as these factors can increase the likelihood of passing on recessive genetic conditions.
Understanding this inheritance pattern is essential for accurate diagnosis and reproductive planning.
Couples who are both known carriers may opt for prenatal genetic testing or preimplantation genetic diagnosis (PGD) during in vitro fertilization to reduce the risk of having an affected child.
Because Wolfram Syndrome is rare, it is often underdiagnosed or misdiagnosed, especially in early stages.
Identifying the genetic mechanism through family history and testing enables early monitoring, timely interventions, and access to multidisciplinary care.
Genetic counseling not only helps affected families understand their risks but also empowers them to make informed decisions for future generation.
Current Research and Future Directions
Recent research has focused on better understanding the genetic pathways involved in Wolfram Syndrome to develop targeted therapies.
For instance, trials exploring the potential of regenerative medicine to replace lost beta cells and protect optic nerve cells are underway.
Gene editing techniques like CRISPR also hold promise for correcting the underlying genetic mutations. However, these are still in experimental stages and not yet available for clinical use.
FAQs on DIDMOAD
Q-1: What does DIDMOAD stand for in Wolfram syndrome explained simply?
A-1: DIDMOAD stands for Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, and Deafness—the four defining features of Wolfram syndrome. This rare genetic disorder affects fewer than 5,000 individuals in the United States, making it difficult to diagnose early. Symptoms do not appear all at once; instead, they develop gradually over time, often beginning with diabetes mellitus in childhood. This staggered presentation is one reason the condition remains underrecognized.
Q-2: Why is DIDMOAD considered a hidden puzzle in Wolfram syndrome diagnosis?
A-2: DIDMOAD is considered a “hidden puzzle” because its symptoms emerge across different body systems over many years. For instance, a child may first be diagnosed with diabetes, while vision and hearing problems may not appear until adolescence. This gap—often spanning 8 to 10 years—leads to fragmented diagnoses. Healthcare providers may treat each symptom separately, delaying recognition of the underlying syndrome.
Q-3: How are diabetes insipidus and diabetes mellitus linked in Wolfram syndrome?
A-3: In Wolfram syndrome, both diabetes insipidus and diabetes mellitus occur due to dysfunction in different regulatory systems of the body. Diabetes mellitus arises from reduced insulin production in the pancreas, while diabetes insipidus results from impaired water balance controlled by the brain. The coexistence of these two conditions is rare in the general population but becomes a hallmark feature in DIDMOAD, seen in a majority of patients as the disease progresses.
Q-4: What are the early symptoms of DIDMOAD syndrome and how can they be identified?
A-4: The earliest symptoms typically include juvenile-onset diabetes mellitus and gradual vision loss caused by optic atrophy, often appearing before the age of 16. Studies show that nearly 90% of patients first present with diabetes. Early identification requires careful monitoring of visual changes in children already diagnosed with diabetes, as this combination strongly संकेत toward Wolfram syndrome.
Q-5: Why is Wolfram syndrome often misdiagnosed as type 1 diabetes?
A-5: Wolfram syndrome is often misdiagnosed as type 1 diabetes because both conditions share early symptoms such as high blood sugar and insulin dependence. However, unlike typical type 1 diabetes, Wolfram syndrome later involves neurological and sensory complications. This overlap leads to misdiagnosis in a significant number of early cases, especially in pediatric populations across the United States.
Q-6: What are the genetic causes of DIDMOAD and the role of WFS1 gene mutation?
A-6: More than 90% of Wolfram syndrome cases are caused by mutations in the WFS1 gene, located on chromosome 4. This gene is responsible for regulating cellular stress and maintaining proper insulin production. When mutated, it disrupts multiple systems in the body, leading to the DIDMOAD symptom cluster. The disorder follows an autosomal recessive inheritance pattern, meaning a child must inherit the mutated gene from both parents.
Q-7: How do optic atrophy and hearing loss develop in Wolfram syndrome patients?
A-7: Optic atrophy develops due to the degeneration of optic nerve fibers, leading to progressive vision loss. Hearing loss, which affects more than 50% of patients, typically begins during adolescence and worsens over time. These symptoms reflect the neurodegenerative nature of the disease, as it gradually damages nerve cells responsible for sensory functions.
Q-8: What makes DIDMOAD a rare and complex neurodegenerative disorder?
A-8: Wolfram syndrome is extremely rare, with an estimated prevalence of about 1 in 100,000 people in North America. Its complexity lies in its multi-system involvement, simultaneously affecting the endocrine system, nervous system, vision, and hearing. This combination of symptoms makes it one of the most challenging genetic disorders to diagnose and manage effectively.
Q-9: What is the life expectancy and progression timeline of Wolfram syndrome (DIDMOAD)?
A-9: Wolfram syndrome is a progressive condition with an average life expectancy of 30 to 40 years. Symptoms worsen over time, often leading to significant neurological complications. The progression typically begins in childhood with diabetes, followed by vision loss in adolescence, and later hearing and neurological decline in early adulthood.
Q-10: What are the latest research and treatment options for DIDMOAD syndrome in the USA?
A-10: Currently, there is no cure for Wolfram syndrome, but research is advancing rapidly. Clinical trials in the United States are exploring gene therapy and treatments aimed at reducing cellular stress. Programs supported by rare disease initiatives have accelerated progress, offering hope to the estimated 3,000 to 5,000 individuals affected nationwide. While treatments remain supportive, early diagnosis and intervention can improve quality of life significantly.
Takeaway: Solving the Puzzle of DIDMOAD
So, why is DIDMOAD considered Wolfram Syndrome’s hidden puzzle?
The complexity lies in its wide range of symptoms and genetic variability, making diagnosis and treatment incredibly challenging.
Prevention is better than cure. So, do make an effort to learn how to lower blood sugar naturally using the best diabetic supplements.
Advances in genetic research and a better understanding of the disease’s pathophysiology are slowly unraveling this puzzle, offering hope for more effective treatments in the future.
References: