How KCNJ11 Mutations Disrupt Beta-Cell Function in Neonates?
- admin
- December 21, 2024
- 9:11 pm
- No Comments
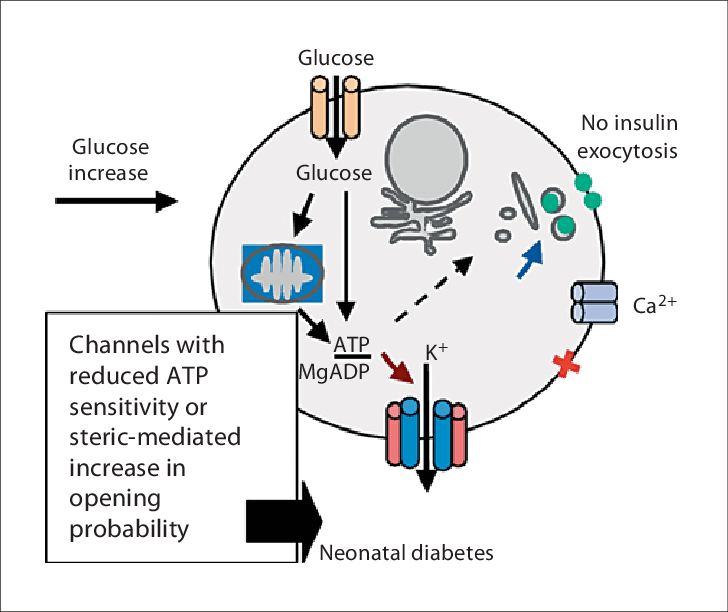
Beta-cell function is crucial for maintaining glucose homeostasis, especially in neonates who are adapting to independent glucose regulation after birth.
The KCNJ11 gene, encoding the Kir6.2 subunit of the ATP-sensitive potassium (KATP) channel, plays a pivotal role in insulin secretion.
However, mutations in KCNJ11 can lead to beta-cell dysfunction, causing neonatal diabetes.
This article by bestdietarysupplementfordiabetics.com delves into how KCNJ11 mutations disrupt beta-cell function in neonates, focusing on the mechanisms involved, real-life examples, and supporting scientific evidence.
Article Index:
- Introduction to KCNJ11 and Its Role in Beta-Cell Function
- Mechanisms of Beta-Cell Dysfunction Due to KCNJ11 Mutations
- Persistent KATP Channel Activity
- Impaired Membrane Depolarization
- Calcium Influx Disruption
- Genetic Mutations in KCNJ11 and Their Clinical Impact
- Gain-of-Function Mutations
- Variability in Mutation Expression
- Real-Life Case Studies
- Case Study 1: Neonatal Diabetes in an Infant with KCNJ11 Mutation
- Case Study 2: Familial Diabetes Linked to KCNJ11 Mutation
- Scientific Evidence Supporting KCNJ11 Mutations and Neonatal Diabetes
- FAQs on KCNJ11 Mutations and Neonatal Diabetes
- Conclusion
Introduction to KCNJ11 and Its Role in Beta-Cell Function
The KCNJ11 gene encodes the Kir6.2 protein, a critical subunit of the ATP-sensitive potassium (KATP) channel in pancreatic beta cells.
This channel serves as a metabolic sensor, linking glucose metabolism to insulin secretion.
Under normal conditions, glucose metabolism raises intracellular ATP levels, closing the KATP channel.
This closure leads to cell membrane depolarization, which triggers the opening of voltage-dependent calcium channels.
The subsequent calcium influx stimulates insulin granule exocytosis, ensuring precise glucose regulation.
Mutations in KCNJ11 disrupt this finely tuned mechanism. Gain-of-function mutations in the Kir6.2 protein prevent the KATP channel from closing in response to ATP, impairing membrane depolarization and blocking calcium entry.
Without sufficient calcium influx, insulin granules cannot be released, leading to persistent hyperglycemia in neonates.
Research published in Nature Genetics (Gloyn et al., 2004) demonstrated that KCNJ11 mutations are a primary cause of neonatal diabetes, emphasizing their critical role in beta-cell dysfunction and glucose imbalance.
Mechanisms of Beta-Cell Dysfunction Due to KCNJ11 Mutations
A quick look at these aspects:
Persistent KATP Channel Activity:
Gain-of-function mutations in KCNJ11 often keep the KATP channel persistently open, even when ATP levels are high.
This prevents the necessary membrane depolarization required for insulin granule exocytosis.
Research in Nature Genetics (Gloyn et al., 2004) demonstrated that such mutations are a primary cause of permanent neonatal diabetes.
Impaired Membrane Depolarization:
Membrane depolarization is a critical step in insulin secretion.
When the KATP channel fails to close, beta cells cannot achieve the voltage change needed to activate voltage-dependent calcium channels.
Without this activation, insulin release is severely compromised, leading to glucose dysregulation.
Calcium Influx Disruption:
Calcium influx is the final trigger for insulin granule exocytosis.
Disruption in the KATP channel’s function due to KCNJ11 mutations prevents this influx, halting the insulin secretion process.
A study in Diabetes (Ashcroft & Rorsman, 2012) elaborated on how KATP channel defects caused by KCNJ11 mutations impair calcium signaling pathways.
Genetic Mutations in KCNJ11 and Their Clinical Impact
Here is how it happens:
Gain-of-Function Mutations:
Mutations in the KCNJ11 gene, such as R201H and V59M, cause gain-of-function effects, resulting in excessive KATP channel activity.
This prevents the channel from closing in response to rising ATP levels during glucose metabolism, thereby disrupting insulin secretion.
Neonates with these mutations often develop permanent neonatal diabetes mellitus (PNDM), characterized by persistent hyperglycemia.
Pharmacological interventions like sulfonylureas, which target and close the KATP channels, have proven effective in restoring partial insulin secretion.
Research in Nature Genetics (Gloyn et al., 2004) demonstrated how sulfonylureas improved blood glucose control in neonates with specific KCNJ11 mutations.
Variability in Mutation Expression:
Interestingly, the clinical presentation of KCNJ11 mutations varies widely, even among individuals with the same mutation.
Some exhibit transient neonatal diabetes mellitus (TNDM) that resolves within months, while others develop PNDM requiring lifelong management.
This variability is influenced by environmental factors, such as neonatal stress, and genetic modifiers, including variations in other genes affecting beta-cell function.
A pivotal study in The Journal of Clinical Investigation (Hattersley et al., 2006) demonstrated how sulfonylureas significantly improved glycemic control in patients with KCNJ11 mutations, underscoring the importance of genotype-specific therapies.
This highlights the role of personalized medicine in managing beta-cell dysfunction caused by KCNJ11 mutations.
Real-Life Case Studies
Below listed are a few examples that we would like to discuss with you:
Ethan’s Journey: Managing Neonatal Diabetes with Genetic Insight
Ethan, a two-month-old infant, exhibited alarming symptoms of persistent hyperglycemia and failure to thrive shortly after birth.
Diagnostic testing pinpointed a V59M mutation in the KCNJ11 gene, a common cause of neonatal diabetes. This mutation disrupted the closure of KATP channels, preventing insulin secretion from beta cells in response to glucose.
Early intervention with oral sulfonylureas, which work by closing KATP channels pharmacologically, partially restored Ethan’s insulin release.
Within weeks of treatment, his blood glucose levels stabilized, and his weight gain improved significantly.
Ethan’s case underscores the transformative potential of genetic testing and targeted therapies in managing neonatal diabetes.
A Familial Link: Diabetes Across Generations
In another case, a family spanning three generations was studied due to a recurring pattern of diabetes.
Genetic analysis revealed an R201H mutation in the KCNJ11 gene shared by the grandfather, mother, and child. Interestingly, their clinical presentations varied widely.
While the grandfather required insulin injection therapy, the mother managed her condition with dietary adjustments, and the child presented with neonatal diabetes.
This variability highlights the concept of variable expressivity, where the same genetic mutation results in differing clinical outcomes depending on environmental factors and other genetic modifiers.
The case emphasizes the importance of genetic counseling in understanding the risks, prognosis, and personalized management strategies for families with KCNJ11 mutations.
Scientific Evidence Supporting KCNJ11 Mutations and Neonatal Diabetes
Here are some of these:
Gloyn et al., 2004 (Nature Genetics):
This groundbreaking study established a direct link between gain-of-function mutations in the KCNJ11 gene and neonatal diabetes. The researchers demonstrated that mutations, such as R201H and V59M, disrupt the sensitivity of the KATP channel to ATP, preventing its closure. This defect impairs insulin secretion, laying the foundation for understanding the genetic basis of neonatal diabetes.
Ashcroft & Rorsman, 2012 (Diabetes):
This research delved into the biophysical mechanisms of KATP channel dysfunction caused by KCNJ11 mutations. The study explained how impaired channel closure affects calcium influx, a critical step for insulin granule exocytosis. These findings clarified the molecular pathways linking KATP channel defects to reduced insulin secretion.
Hattersley et al., 2006 (The New England Journal of Medicine):
This study focused on the therapeutic role of sulfonylureas, a drug class that closes KATP channels pharmacologically. It showed that neonates with KCNJ11 mutations experienced improved glucose regulation and reduced reliance on insulin therapy when treated with sulfonylureas, revolutionizing treatment strategies for neonatal diabetes.
Stanley et al., 2016 (Pediatric Diabetes):
The study highlighted the clinical variability of KCNJ11 mutations. Some individuals with the same mutation presented with transient neonatal diabetes, while others developed permanent diabetes.
The research emphasized the role of genetic modifiers and environmental factors in influencing outcomes.
Edghill et al., 2007 (Human Molecular Genetics):
This study demonstrated that KCNJ11 mutations not only impair insulin secretion but also induce beta-cell apoptosis. The resulting loss of beta-cell mass exacerbates insulin deficits, contributing to the progression of neonatal diabetes.
FAQs on KCNJ11 Mutations and Neonatal Diabetes
Q-1: What exactly does a KCNJ11 mutation do to a newborn’s β-cells?
A-1: KCNJ11 encodes Kir6.2, the pore-forming half of the β-cell KATP channel. Activating mutations make the channel less sensitive to ATP, so it stays too open even when glucose is high. Without KATP closure, the membrane won’t depolarize, calcium doesn’t surge, and insulin granules aren’t released. Clinically, that looks like hyperglycemia within the first months of life despite otherwise healthy-appearing infants, often with preserved C-peptide but inappropriately low insulin for the glucose level.
Q-2: Why do some KCNJ11 mutations also cause neurological symptoms (DEND) along with diabetes?
A-2: The same Kir6.2/SUR1 KATP architecture found in β-cells is also present in neurons. Strongly activating variants keep neuronal KATP channels open, dampening excitability during development. Depending on mutation strength and distribution, this can produce developmental delay, epilepsy, and hypotonia—hence DEND (Developmental delay, Epilepsy, and Neonatal Diabetes). Milder variants may cause diabetes alone; more disruptive ones affect both pancreatic and neural circuits.
Q-3: How does this channel behavior shape treatment—why can sulfonylureas replace insulin in many cases?
A-3: Sulfonylureas bind SUR1 (the regulatory KATP subunit) and force channels to close, bypassing the defective ATP sensing. Once KATP closes, β-cells depolarize and secrete insulin in a glucose-responsive fashion. Many infants—and even older children misclassified as type 1—can transition from injections to oral sulfonylureas with improved glucose profiles. Dose needs vary by variant; some require higher doses or partial insulin support, but overall responsiveness is high when started early.
Q-4: What molecular details explain how specific KCNJ11 variants disrupt glucose sensing?
A-4: Kir6.2 gating is tuned by ATP (inhibitory) and membrane lipids like PIP₂ (activating), coordinated through interfaces with SUR1. Mutations that strengthen open-state stabilization or weaken ATP inhibition shift the channel toward persistent openness. Others alter subunit coupling or trafficking, effectively raising the glucose threshold for insulin release. Small amino-acid substitutions at these interfaces can magnify currents dramatically—enough to block first-phase insulin even at neonatal glucose loads.
Q-5: Which clinical clues should prompt genetic testing for KCNJ11, and what’s the outlook?
A-5: Red flags include hyperglycemia before 6 months of age, negative islet autoantibodies, detectable C-peptide, and a family history compatible with autosomal dominance. Rapid genetic confirmation directs therapy toward sulfonylureas, which can normalize or markedly improve glycemia and may benefit neurodevelopment when instituted early. Long-term prognosis depends on mutation severity: some children have isolated diabetes with excellent control; others with DEND need multidisciplinary care for seizures, tone, and developmental support alongside glucose management.
Conclusion
Mutations in the KCNJ11 gene, which encodes the Kir6.2 protein of the KATP channel, significantly impair beta-cell function by disrupting ATP sensitivity.
Under normal conditions, the KATP channel closes in response to increased ATP levels, leading to membrane depolarization, calcium influx, and insulin secretion.
However, gain-of-function mutations, such as R201H and V59M, keep the channel persistently open, preventing insulin release and causing persistent hyperglycemia—a hallmark of neonatal diabetes.
This dysfunction highlights the complex molecular basis of neonatal diabetes, emphasizing the importance of early genetic diagnosis and targeted treatments like sulfonylureas, which pharmacologically close KATP channels to restore partial insulin secretion.
Advances in research, including studies like those by Gloyn et al. (2004) and Hattersley et al. (2006), have revolutionized treatment strategies and improved outcomes for affected neonates.
Scientific progress in understanding KCNJ11 mutations continues to provide insights into beta-cell dysfunction and its management.
These advancements not only pave the way for personalized therapies but also offer hope for developing innovative treatments to address neonatal diabetes more effectively.
References: