Neurodegeneration in Wolfram Syndrome (WS) is a complex, progressive condition that impacts multiple systems of the body.
While Wolfram Syndrome is primarily known for its acronym DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness), neurodegeneration underpins many of the syndrome’s symptoms.
This article by bestdietarysupplementfordiabetics.com explores the mechanisms, progression, and impact of neurodegeneration in WS, supported by scientific evidence and real-life examples to provide a comprehensive understanding of this rare disorder.
Article Index:
- Overview of Wolfram Syndrome and Neurodegeneration
- Role of WFS1 Gene in Neurodegeneration
- Key Symptoms Linked to Neurodegeneration
- Optic Atrophy
- Hearing Loss
- Brainstem and Cerebellar Degeneration
- Mechanisms of Neurodegeneration in Wolfram Syndrome
- Endoplasmic Reticulum (ER) Stress
- Calcium Dysregulation
- Mitochondrial Dysfunction
- Progression of Neurodegenerative Symptoms
- Scientific Evidence Supporting Neurodegenerative Mechanisms in WS
- Real-Life Examples of Neurodegeneration in Wolfram Syndrome
- FAQs on Neurodegeneration in Wolfram Syndrome
- Current Research and Diagnostic Approaches
- Conclusion
Overview of Wolfram Syndrome and Neurodegeneration
Wolfram Syndrome is a rare genetic disorder primarily caused by mutations in the WFS1 gene, which plays a crucial role in cellular homeostasis and endoplasmic reticulum (ER) stress regulation.
These mutations disrupt normal cellular processes, leading to progressive damage to the nervous system and widespread neurodegeneration.
Unlike other neurodegenerative conditions that typically manifest in older individuals, Wolfram Syndrome often begins in childhood, with symptoms progressively worsening throughout life.
Early manifestations include optic atrophy and diabetes mellitus, followed by neurological complications such as ataxia, cognitive decline, and psychiatric issues, which significantly contribute to the disease’s burden.
These neurological symptoms arise from damage to the brainstem and cerebellum, areas essential for motor coordination, speech, and autonomic functions.
The progressive nature of the disorder underscores the importance of early diagnosis and comprehensive management strategies to address its complex, multisystem challenges.
Role of WFS1 Gene in Neurodegeneration
The WFS1 gene encodes wolframin, a vital protein responsible for maintaining calcium homeostasis and regulating endoplasmic reticulum (ER) stress.
Wolframin plays a critical role in ensuring proper protein folding and intracellular calcium signaling, both essential for cellular stability and function.
Mutations in the WFS1 gene disrupt these processes, leading to cellular stress and increased vulnerability to damage.
This dysfunction is particularly pronounced in neurons within the brainstem and cerebellum, which have high metabolic demands and are highly sensitive to calcium imbalances.
These regions are integral to motor coordination and autonomic functions, making them primary targets for neurodegeneration in Wolfram Syndrome.
Scientific Insight:
Research published in The American Journal of Human Genetics (2010) identified over 200 mutations in the WFS1 gene, emphasizing its genetic heterogeneity.
The study also established a correlation between specific mutations and the severity of neurodegenerative symptoms, underscoring the critical role of wolframin in neuronal health and disease progression.
Key Symptoms Linked to Neurodegeneration
Optic Atrophy:
Optic atrophy is often one of the earliest signs of Wolfram Syndrome (WS).
It involves the progressive loss of optic nerve fibers, which compromises the transmission of visual information from the eyes to the brain.
Patients initially experience mild vision impairment, which can progress to severe vision loss or complete blindness over time.
Regular ophthalmological evaluations are critical for monitoring and managing this symptom.
Hearing Loss:
Sensorineural hearing loss is another hallmark of WS, arising from damage to auditory neurons.
Typically beginning in adolescence, this type of hearing loss gradually worsens with age, significantly impacting communication and quality of life.
Early audiological interventions, including hearing aids, can help manage this progression.
Brainstem and Cerebellar Degeneration:
Neurological symptoms such as ataxia, dysarthria, and balance issues result from the degeneration of the brainstem and cerebellum—regions essential for motor coordination, speech, and balance.
These symptoms often emerge in adulthood and contribute to the progressive loss of independence in WS patients.
Physical therapy and assistive devices are frequently employed to address these challenges and improve mobility and coordination.
Mechanisms of Neurodegeneration in Wolfram Syndrome
A quick look at this aspect:
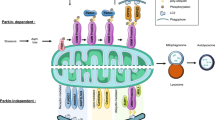
Endoplasmic Reticulum (ER) Stress:
ER stress is a hallmark of neurodegeneration in Wolfram Syndrome (WS).
Mutations in the WFS1 gene disrupt the ER’s ability to process and fold proteins, causing an accumulation of misfolded proteins and triggering cellular stress responses.
Persistent ER stress activates pathways leading to apoptosis, resulting in the progressive loss of neurons critical for various functions.
Scientific Evidence:
A study published in Cell Death & Disease (2015) demonstrated that therapeutic strategies aimed at reducing ER stress in cellular models of WS significantly mitigated neuronal loss, highlighting its central role in the disease pathology.
Calcium Dysregulation:
Wolframin, the protein encoded by WFS1, is essential for maintaining calcium homeostasis.
Dysregulated calcium levels disrupt neuronal signaling pathways and impair the generation of cellular energy.
This imbalance further exacerbates neurodegeneration by weakening neuronal functionality and resilience.
Mitochondrial Dysfunction:
Calcium dysregulation directly affects mitochondrial performance, a critical component of energy production in neurons.
Impaired mitochondrial function leads to reduced ATP synthesis and increased oxidative stress, compounding neuronal damage and accelerating neurodegenerative processes in WS.
Together, these interconnected mechanisms create a cascade that drives the progression of neurological symptoms.
Progression of Neurodegenerative Symptoms
Neurodegenerative symptoms in Wolfram Syndrome (WS) follow a distinct and progressive pattern, impacting patients at various life stages:
- Childhood: The earliest signs often include optic atrophy, leading to vision loss, and diabetes mellitus, characterized by high blood sugar levels requiring early management.
- Adolescence: Symptoms such as sensorineural hearing loss and coordination difficulties, including issues with balance and motor skills, typically emerge, further affecting quality of life.
- Adulthood: Neurological complications intensify, presenting as ataxia, cognitive decline, and psychiatric disorders like anxiety or depression, which significantly impair independence and daily functioning.
This progression underscores the importance of early diagnosis and multidisciplinary care to address evolving symptoms and improve patient outcomes.
What does Science Have to Say?
- Hershey et al. (2012): This pivotal study emphasized endoplasmic reticulum (ER) stress as a central mechanism in the neurodegeneration observed in Wolfram Syndrome (WS). The research provided insights into how disrupted protein folding contributes to neuronal loss, paving the way for targeted therapeutic strategies.
- Riano et al. (2019): Focused on mitochondrial dysfunction, this study linked impaired energy production and increased oxidative stress to the neuronal degeneration characteristic of WS. The findings underscored the critical role of mitochondria in maintaining neuronal health.
- Barrett et al. (2018): Investigated potential therapeutic interventions aimed at restoring wolframin function. This research explored the use of chemical chaperones and other agents to reduce ER stress and improve cellular resilience, offering hope for mitigating neurodegenerative progression in WS patients.
Case Study: Emily’s Vision Loss
Emily, diagnosed with Wolfram Syndrome at the age of 8, initially presented with diabetes mellitus and mild vision problems.
Over time, progressive optic atrophy significantly deteriorated her eyesight, leaving her legally blind by her teenage years.
This loss of vision not only affected her academic performance but also her emotional well-being.
Genetic testing confirmed WFS1 mutations, enabling her healthcare team to develop a tailored management plan.
Emily’s case underscores the importance of early diagnosis and intervention, including routine ophthalmological evaluations and access to assistive technologies like screen readers and magnification devices.
Case Study: Michael’s Balance Issues
Michael, a 22-year-old living with WS, began experiencing coordination difficulties and frequent falls in his late teens.
His symptoms were attributed to cerebellar degeneration, a hallmark of the neurodegenerative aspects of WS. The ataxia significantly impacted his ability to perform daily activities and maintain independence.
Physical therapy sessions focused on improving his strength and balance, while assistive devices such as walkers helped him navigate his environment more safely.
Michael’s journey highlights the progressive nature of neurological symptoms in WS and the importance of multidisciplinary care to address mobility challenges and maintain quality of life.
Current Research and Diagnostic Approaches
Advances in genetic testing have significantly enhanced the accuracy of Wolfram Syndrome (WS) diagnosis, allowing for earlier detection and targeted intervention.
Nonetheless, Wolfram Syndrome is still a mystery.
Techniques such as whole-exome sequencing and next-generation sequencing are instrumental in identifying WFS1 mutations, providing detailed insights into the genetic underpinnings of the disorder.
These genetic tools also facilitate the identification of carriers within families, aiding in genetic counseling and risk assessment.
Additionally, imaging modalities like MRI play a crucial role in diagnosing WS, revealing brainstem and cerebellar atrophy linked to the neurodegenerative aspects of the condition.
MRI findings often correlate with disease progression, offering a valuable tool for monitoring.
Emerging Research:
Efforts to mitigate ER stress and restore calcium homeostasis are at the forefront of WS therapeutic development.
Promising approaches include the use of chemical chaperones to stabilize protein folding and reduce cellular stress.
Preclinical studies, such as those published in Cell Death & Disease (2019), have demonstrated the potential of these treatments to slow neurodegeneration and preserve neuronal function.
Ongoing clinical trials aim to translate these findings into viable therapies, offering hope for improved management and outcomes in WS patients.

FAQs on Neurodegeneration in Wolfram Syndrome
Q1: What is the role of the WFS1 gene in neurodegeneration associated with Wolfram syndrome?
A1: The WFS1 gene produces a protein called wolframin that is important for maintaining the health of the endoplasmic reticulum (ER) in cells. Mutations in this gene cause ER stress and disrupt calcium balance and mitochondrial function, triggering neurodegenerative processes in Wolfram syndrome.
Q2: How does Wolfram syndrome affect myelin development in the nervous system?
A2: People with Wolfram syndrome often experience defective myelin development, leading to reduced integrity of the myelin sheath around nerve fibers, especially in the optic tracts. This impaired myelination contributes to the neurological symptoms seen in the condition.
Q3: What are the early signs of neurodegeneration in Wolfram syndrome?
A3: Early signs include difficulties with balance and coordination, often presenting as ataxia in early adulthood. Brainstem atrophy, particularly in the ventral pons region, is also common and can result in complications like central apnea.
Q4: How does Wolfram syndrome impact white matter in the brain?
A4: Neuroimaging shows that Wolfram syndrome causes a decrease in white matter volume, particularly in areas such as the basilar pons, cerebellar white matter, and visual cortex. The extent of white matter loss correlates with the severity of neurological impairment.
Q5: Are there any therapeutic approaches targeting neurodegeneration in Wolfram syndrome?
A5: Experimental treatments combining liraglutide, a GLP-1 receptor agonist, with a neurotrophic agent called 7,8-DHF have shown promise. This combination may help protect neurons and stabilize or improve vision in animal models of Wolfram syndrome.
Q6: What is the significance of oligodendrocytes in Wolfram syndrome-related neurodegeneration?
A6: Oligodendrocytes, which are responsible for forming myelin sheaths around neurons, express the WFS1 protein during early brain development. Their dysfunction due to WFS1 mutations is believed to play a key role in the neurodegenerative process of Wolfram syndrome.
Conclusion
Neurodegeneration in Wolfram Syndrome is a multifaceted process involving ER stress, calcium dysregulation, and mitochondrial dysfunction.
Understanding these mechanisms is crucial for developing effective interventions. While current treatments focus on managing symptoms, ongoing research offers hope for therapies that target the root causes of neurodegeneration.
Real-life experiences underscore the profound impact of WS on patients’ lives, highlighting the importance of continued research and comprehensive care.
References:
admin
All Posts