Genetic Basis of GCK-Related Neonatal Diabetes
- admin
- December 28, 2024
- 12:01 pm
- No Comments
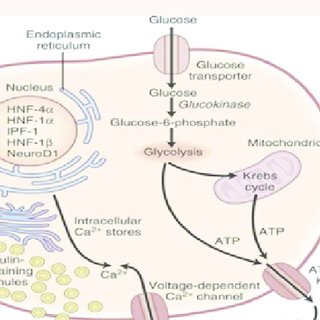
Neonatal diabetes is a rare condition characterized by hyperglycemia within the first six months of life, often caused by mutations in specific genes regulating insulin production.
One such gene is GCK (Glucokinase), which plays a pivotal role in glucose sensing and metabolism in pancreatic beta cells.
GCK-related neonatal diabetes results from either activating or inactivating mutations, disrupting the delicate balance of glucose homeostasis.
In this article, we will explore the genetic mechanisms underlying GCK-related neonatal diabetes, supported by scientific evidence and real-life examples, to understand how this rare form of diabetes manifests.
Article Index:
- Introduction to GCK and Its Role in Glucose Metabolism
- How Mutations in GCK Lead to Neonatal Diabetes
- Types of GCK Mutations and Their Effects
- Inheritance Patterns in GCK-Related Neonatal Diabetes
- Clinical Presentation and Diagnosis
- Scientific Evidence Supporting the Genetic Basis
- Real-Life Examples of GCK Mutations in Neonatal Diabetes
- Conclusion: The Critical Role of GCK in Neonatal Diabetes
Introduction to GCK and Its Role in Glucose Metabolism
The GCK gene encodes glucokinase, a critical enzyme responsible for regulating glucose metabolism in the body.
Glucokinase functions as a glucose sensor, particularly in pancreatic beta cells, where it plays a pivotal role in controlling insulin secretion in response to varying blood glucose levels.
The enzyme catalyzes the phosphorylation of glucose to glucose-6-phosphate, marking the first and essential step in glycolysis.
This process not only initiates glucose metabolism but also helps beta cells determine when to release insulin by setting the glucose threshold for secretion.
Mutations in the GCK gene disrupt this highly regulated system, either enhancing or impairing the enzyme’s activity.
Such disruptions lead to abnormal glucose regulation, causing conditions like persistent hyperglycemia or hypoinsulinemia.
The enzyme’s central role highlights the importance of GCK in maintaining glucose homeostasis, and even minor mutations can have profound metabolic consequences.
How Mutations in GCK Lead to Neonatal Diabetes
Mutations in the GCK gene significantly impact the enzyme’s ability to respond to glucose, disrupting its function as a key regulator of glucose metabolism and insulin secretion.
These mutations can lead to either excessive or insufficient insulin release, depending on their nature.
Activating mutations, which lower the glucose threshold for insulin secretion, are often responsible for neonatal diabetes.
In these cases, beta cells release insulin even at low glucose levels, resulting in persistent hyperglycemia.
Conversely, inactivating mutations reduce glucokinase activity, causing beta cells to inadequately respond to rising glucose levels, leading to hypoinsulinemia and poor glucose regulation.
This imbalance underscores the enzyme’s crucial role in maintaining glucose homeostasis and highlights the delicate interplay between glucokinase function and metabolic health.
Even subtle changes in GCK activity due to mutations can have profound effects on glucose sensing and insulin release, emphasizing the importance of precise regulation.
Types of GCK Mutations and Their Effects
Mutations in the GCK gene are categorized into two main types: activating and inactivating mutations, each producing distinct physiological effects:
- Activating Mutations: These rare mutations enhance the enzyme’s activity, lowering the glucose threshold required for insulin secretion. This results in hyperglycemia, as insulin release occurs even at low blood glucose levels.
- Example Mutation: A179E alters the active site of glucokinase, increasing its affinity for glucose and over-activating its function.
- Inactivating Mutations: More frequently associated with GCK-related diabetes, these mutations impair glucokinase activity. This impairment prevents beta cells from responding adequately to rising blood glucose levels, leading to hypoinsulinemia and poor glucose regulation.
- Example Mutation: G261R disrupts the enzyme’s structural integrity, significantly reducing its catalytic efficiency and ability to phosphorylate glucose.
These mutations highlight the delicate balance glucokinase maintains in regulating glucose metabolism and insulin secretion.
Understanding these mutations provides critical insights into the mechanisms driving GCK-related neonatal diabetes.
Inheritance Patterns in GCK-Related Neonatal Diabetes
GCK-related neonatal diabetes is most commonly inherited in an autosomal dominant manner.
In this pattern, a single copy of the mutated GCK gene is sufficient to cause the condition, often leading to mild-to-moderate hyperglycemia.
However, rare cases of autosomal recessive inheritance have also been documented, requiring both copies of the gene to carry mutations for the condition to manifest.
Autosomal recessive forms are typically associated with more severe phenotypes, including profound glucose dysregulation.
Understanding the inheritance pattern is essential for accurate diagnosis and management. Genetic testing plays a pivotal role in identifying the specific mutation and its inheritance mode.
This information is invaluable for family planning, allowing parents to assess potential risks for future offspring.
It also aids in tailoring treatment strategies, ensuring optimal care for individuals affected by this rare genetic condition.
Clinical Presentation and Diagnosis
The clinical presentation of GCK-related neonatal diabetes typically includes persistent hyperglycemia, which becomes apparent within the first six months of life.
Unlike type 1 diabetes, this condition does not involve ketoacidosis, making it a distinctive diagnostic feature.
Symptoms are generally mild and may include increased thirst (polydipsia) and frequent urination (polyuria).
These subtle manifestations can sometimes delay diagnosis, emphasizing the need for awareness among healthcare providers.
Diagnostic Tools:
- Genetic Testing: This is the gold standard for diagnosing GCK-related neonatal diabetes. It helps identify specific mutations in the GCK gene, confirming the genetic basis of the condition.
- Glucose Tolerance Tests: These assess beta cell functionality and provide additional insights into the extent of glucose dysregulation.
Early diagnosis through genetic screening is crucial. It prevents potential complications, facilitates personalized management strategies, and offers valuable insights for family planning and risk assessment.
Scientific Evidence Supporting the Genetic Basis
Numerous studies have established the genetic basis of GCK-related neonatal diabetes:
- Gloyn et al., 2004: Published in Diabetologia, this study identified GCK mutations as a primary cause of neonatal diabetes, emphasizing the importance of glucokinase in glucose sensing.
- Murphy et al., 2008: Research in Human Mutation highlighted the structural and functional impact of various GCK mutations, correlating them with clinical outcomes.
- Hattersley et al., 2006: This study in The Lancet demonstrated that targeted genetic testing for GCK mutations improves diagnostic accuracy and management of neonatal diabetes.
GCK Mutations in Neonatal Diabetes: Emma’s Diagnosis
Emma, a two-month-old infant, was brought to the hospital with persistent hyperglycemia.
She exhibited mild symptoms of increased thirst and frequent urination but showed no signs of ketoacidosis, differentiating her condition from type 1 diabetes.
Genetic testing identified an A179E mutation in her GCK gene, confirming the diagnosis of neonatal diabetes. This mutation enhanced the enzyme’s glucose affinity, leading to dysregulated insulin secretion.
Early diagnosis allowed her pediatrician to tailor her treatment, which included regular glucose monitoring and dietary adjustments.
Her glucose levels stabilized within weeks, enabling her to thrive without complications.
This case highlights the importance of genetic screening for early intervention, as noted in a study by Gloyn et al. (2004) in Diabetologia, which underscores the utility of genetic testing in identifying specific GCK mutations.
GCK Mutations in Neonatal Diabetes: John’s Family History
John, a five-year-old boy, experienced mild hyperglycemia during a routine health check.
His father, who had a history of diabetes diagnosed in adolescence, underwent genetic testing, revealing a G261R mutation in the GCK gene.
Subsequent testing confirmed the same mutation in John, illustrating an autosomal dominant inheritance pattern.
The mutation impaired glucokinase’s catalytic efficiency, resulting in insufficient insulin secretion.
Studies, including one by Hattersley et al. (2006) in The Lancet, emphasize the role of autosomal dominant mutations in familial cases of GCK-related diabetes.
This case underscores the importance of family history in diagnosing hereditary forms of neonatal diabetes, allowing for proactive management strategies across generations.
The Critical Role of GCK in Neonatal Diabetes
GCK-related neonatal diabetes highlights the critical need to understand the genetic underpinnings of rare metabolic disorders.
Mutations in the GCK gene, which encodes glucokinase, disrupt its role as a glucose sensor in pancreatic beta cells.
This disruption leads to impaired insulin secretion and persistent hyperglycemia, distinguishing it from other forms of diabetes.
Advances in genetic testing have revolutionized the diagnosis of GCK-related neonatal diabetes, enabling healthcare providers to identify specific mutations accurately and tailor treatment strategies.
Research has also shed light on the diverse clinical presentations of GCK mutations, ranging from mild hyperglycemia to more complex metabolic conditions.
Studies such as those by Gloyn et al. (2004) and Hattersley et al. (2006) emphasize the role of genetic screening in improving outcomes and guiding family planning.
As scientific understanding of GCK mutations continues to evolve, there is growing potential to enhance management strategies and explore preventive measures for at-risk families.
References: