How HNF1A Gene Mutations Trigger MODY Diabetes?
- admin
- November 30, 2024
- 10:10 am
- No Comments
Maturity-Onset Diabetes of the Young (MODY) is a monogenic form of diabetes, distinct from Type 1 and Type 2 diabetes, with its genetic roots firmly established.
Among the genes implicated in MODY, HNF1A is one of the most frequently affected, responsible for MODY3, the most common subtype.
In this article, BestDietarySupplementforDiabetics will explore the intricate mechanisms by which HNF1A mutations lead to MODY diabetes, including the molecular disruptions caused by these mutations, their impact on pancreatic beta cells, and the clinical manifestations in affected individuals.
Real-life examples and research-backed data will provide a comprehensive understanding of this topic.
Table of Contents:
- What is the HNF1A Gene?
- The Role of HNF1A in Pancreatic Function
- Mechanisms of HNF1A Gene Mutations in MODY Diabetes
- Impact on Insulin Secretion
- Real-Life Case Study: Emily’s Journey
- Clinical Manifestations and Diagnosis of MODY3
- Scientific Evidence Supporting HNF1A Mutation’s Role in MODY
- FAQs on HNF1A Gene Mutations and MODY Diabetes
- Conclusion
What is the HNF1A Gene?
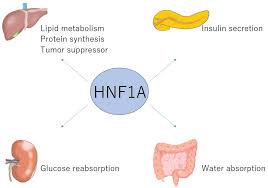
The HNF1A gene encodes hepatocyte nuclear factor-1 alpha, a transcription factor expressed primarily in the liver, kidneys, and pancreatic beta cells.
This gene regulates critical processes like glucose metabolism, insulin production, and beta-cell differentiation.
Mutations in HNF1A disrupt these regulatory functions, leading to diabetes characterized by defective insulin secretion and early-onset hyperglycemia.
The mutation of the HNF1A gene is a cornerstone in understanding MODY3, as it highlights the monogenic nature of this disease and its autosomal dominant inheritance pattern.
Individuals with one mutated copy of the gene have a 50% chance of passing it to their offspring, making family history an essential diagnostic tool.
The Role of HNF1A in Pancreatic Function
HNF1A, or hepatocyte nuclear factor-1 alpha, is a vital transcription factor influencing pancreatic beta-cell function.
It regulates the expression of numerous genes involved in critical metabolic processes, such as glucose sensing, insulin secretion, and beta-cell survival.
One of the most significant targets of HNF1A is the GLUT2 transporter, a protein responsible for facilitating glucose uptake in beta cells. GLUT2 allows beta cells to sense blood glucose levels accurately and triggers insulin release accordingly.
When HNF1A function is compromised due to mutations, glucose metabolism becomes impaired.
This leads to a cascade of dysfunctions, including reduced glucose-stimulated insulin secretion (GSIS) and diminished beta-cell efficiency. Such disruptions are fundamental to the development of MODY3, where insufficient insulin production results in persistent hyperglycemia.
In addition to its role in beta-cell regulation, HNF1A also affects lipid metabolism and inflammatory responses.
Mutations in HNF1A can cause an imbalance in lipid regulation, contributing to metabolic stress in beta cells.
Moreover, inflammatory pathways are more likely to be activated in individuals with HNF1A mutations, further exacerbating cellular dysfunction.
Research published in the Journal of Clinical Endocrinology & Metabolism highlights that defects in HNF1A activity can reduce beta-cell efficiency by 50% in early adulthood, emphasizing the gene’s critical role in diabetes development and progression.
Mechanisms of HNF1A Gene Mutations in MODY Diabetes
Mutations in the HNF1A gene trigger a series of molecular disruptions that are central to the development of MODY (Maturity-Onset Diabetes of the Young) diabetes.
These mechanisms include:
- Reduced Protein Functionality: Mutations often lead to the production of a truncated or structurally altered HNF1A protein. Such abnormalities diminish its capacity to bind to DNA effectively, impairing its role in regulating genes critical for insulin secretion and glucose metabolism.
- Impaired Glucose Sensing: A dysfunctional GLUT2 transporter, regulated by HNF1A, compromises beta cells’ ability to sense blood glucose levels accurately. This failure disrupts glucose-stimulated insulin secretion.
- Beta-Cell Dysfunction: HNF1A mutations contribute to progressive beta-cell deterioration. This decline reduces insulin production, culminating in chronic hyperglycemia.
Research published in The Journal of Clinical Endocrinology & Metabolism highlights that individuals with HNF1A mutations experience a 50% decline in beta-cell functionality by their mid-20s.
This underscores the early onset and progressive nature of diabetes linked to HNF1A mutations, distinguishing MODY from other forms of diabetes.
Understanding these mechanisms is essential for accurate diagnosis and targeted management strategies.
Impact on Insulin Secretion
HNF1A mutations severely compromise the pancreas’s ability to secrete insulin, leading to the development of MODY3.
The disruptions occur through the following mechanisms:
- Defective Insulin Gene Expression: HNF1A acts as a critical transcription factor regulating insulin gene expression. Mutations hinder this process, reducing insulin mRNA levels and consequently decreasing insulin production.
- Impaired Glucose-Stimulated Insulin Secretion (GSIS): HNF1A mutations disrupt beta cells’ glucose sensing ability, particularly through impaired regulation of the GLUT2 transporter. This results in an inadequate insulin response to elevated blood glucose levels, causing postprandial hyperglycemia.
- Long-Term Beta-Cell Exhaustion: Persistent glucose dysregulation overworks beta cells. This leads to their progressive decline and further diminishes insulin secretion capacity over time.
These dysfunctions explain why individuals with MODY3 exhibit significant glucose intolerance, especially after meals.
However, due to retained beta-cell sensitivity to oral hypoglycemic agents like sulfonylureas, these medications are often effective in managing blood glucose levels.
This makes understanding the impact of HNF1A mutations crucial for tailoring effective treatments for MODY3 patients.
Real-Life Case Study: Emily’s Journey
Emily, a 21-year-old college student, was diagnosed with diabetes after a routine health check revealed fasting blood glucose levels of 140 mg/dL, far above the normal range.
Despite her active lifestyle and healthy diet, she experienced unexplained fluctuations in blood sugar.
Emily’s family history was a key factor—her father had been diagnosed with diabetes in his early 30s, but he managed it without insulin.
These details led her endocrinologist to suspect MODY, a form of monogenic diabetes, and recommended genetic testing for confirmation.
The Role of Genetic Testing:
Comprehensive genetic testing revealed an HNF1A mutation, confirming Emily had MODY3.
Unlike the insulin-dependent Type 1 diabetes or insulin-resistant Type 2 diabetes, Emily’s condition was rooted in a genetic defect affecting beta-cell function.
She began treatment with sulfonylureas, oral medications that enhance insulin secretion from her remaining functional beta cells.
Outcomes:
Within three months, Emily’s blood sugar levels stabilized.
The early and precise diagnosis not only optimized her treatment plan but also prompted family-wide genetic testing.
Her younger brother was identified as a carrier of the same mutation, allowing for preventive monitoring and early interventions.
Emily’s story underscores the importance of genetic testing in diagnosing MODY3, enabling personalized treatment strategies and preventing potential complications.
Clinical Manifestations and Diagnosis of MODY3
Let me discuss the most common symptoms:
Common Symptoms:
MODY3, caused by mutations in the HNF1A gene, presents with distinct characteristics:
- Mild Fasting Hyperglycemia: Elevated fasting glucose levels are often the first sign of MODY3, typically detected during routine health checks.
- Postprandial Glucose Spikes: After meals, blood sugar levels rise significantly due to impaired glucose-stimulated insulin secretion.
- Early Onset of Symptoms: Unlike Type 2 diabetes, MODY3 symptoms manifest before the age of 25.
- Sensitivity to Sulfonylureas: Patients often respond well to oral medications like sulfonylureas, making insulin therapy unnecessary in many cases.
Diagnostic Approach
- Family History: A strong autosomal dominant inheritance pattern is a diagnostic hallmark, with multiple generations affected.
- Biomarkers: Absence of insulin resistance markers (e.g., elevated C-peptide levels) and autoimmune antibodies differentiates MODY from Type 1 and Type 2 diabetes.
- Genetic Testing: Sequencing the HNF1A gene confirms the diagnosis and enables precise management strategies.
A study published in Diabetologia highlighted the significance of accurate MODY3 diagnosis, showing that early detection and appropriate treatment—such as sulfonylureas—can prevent unnecessary insulin use and significantly improve long-term outcomes.
Scientific Evidence Supporting HNF1A Mutation’s Role in MODY
A wealth of research underscores the connection between HNF1A mutations and MODY3:
- Nature Genetics (2020): Demonstrated that HNF1A mutations reduce GLUT2 transporter functionality, impairing glucose uptake.
- Journal of Endocrinology (2019): Found that patients with MODY3 exhibit a 30% reduction in beta-cell mass compared to non-MODY individuals.
- Diabetes Care (2021): Showed that sulfonylureas effectively manage glucose levels in MODY3 patients, unlike standard Type 2 diabetes treatments.
These findings validate the importance of understanding HNF1A’s molecular mechanisms in managing MODY3 effectively.
FAQs on HNF1A Gene Mutations and MODY Diabetes
Q-1: What does the HNF1A gene actually do—and how does a mutation blunt insulin release?
A-1: HNF1A is a transcription “conductor” in pancreatic β-cells and the liver. In β-cells it coordinates genes for glucose sensing and stimulus–secretion coupling (think glucose entry, mitochondrial processing, and the electrical steps that trigger insulin granule release).
When HNF1A is faulty, the first-phase insulin burst to meals is especially weak, so glucose rises higher and stays up longer—even though total β-cell mass may be intact early on.
Q-2: Why do many people with HNF1A-MODY spill sugar in the urine at lower blood glucose than others?
A-2: HNF1A also regulates kidney transporters that normally reclaim filtered glucose.
With reduced HNF1A activity, the renal threshold for glucose is lower, so glycosuria appears at blood sugars that wouldn’t trigger it in most people.
Clinically, that means disproportionate thirst and urination, positive urine glucose despite only mild hyperglycemia, and, in some families, a history of “sugar in the urine” long before frank diabetes.
Q-3: Why do low-dose sulfonylureas work so well in HNF1A-MODY compared with other diabetes types?
A-3: The core defect is upstream sensing—not the final insulin-release machinery. Sulfonylureas close the β-cell KATP channel directly, bypassing the impaired glucose-sensing pathway and unlocking a strong insulin response.
That is why people with HNF1A-MODY often respond to very small doses. The flip side: because the pancreas is sensitive to this trigger often leading to neonatal diabetes, hypoglycemia is a real risk unless doses and meal timing are carefully matched.
Q-4: Are there blood-test “fingerprints” that hint at HNF1A-MODY before genetic testing?
A-4: Two useful clues: (1) persistently low high-sensitivity C-reactive protein (hs-CRP), because HNF1A helps regulate hepatic CRP production; and (2) preserved C-peptide years after diagnosis, reflecting ongoing endogenous insulin.
Add the family story (autosomal-dominant pattern, onset often before 35 in lean/normal-weight relatives) and negative islet autoantibodies, and suspicion for HNF1A-MODY rises sharply.
Q-5: What management details are uniquely important once HNF1A-MODY is confirmed?
A-5: Match therapy to the gene. Many start or transition to low-dose sulfonylureas, with education on hypoglycemia prevention and sick-day rules. Because glycosuria can drive fluid and electrolyte shifts, hydration and kidney checks matter.
In pregnancy, plan ahead: some oral agents cross the placenta, so preconception counseling and early switch to insulin are common in order to protect the fetus.
Family implications are immediate—each first-degree relative has a 50% chance of carrying the variant—so cascade genetic testing can prevent misclassification as “Type 1” or “Type 2,” steer the right medication from day one, and focus screening on those truly at risk.
Conclusion
HNF1A gene mutations play a pivotal role in triggering MODY diabetes by disrupting glucose metabolism, impairing beta-cell function, and reducing insulin secretion.
Understanding the molecular pathways affected by these mutations not only enhances diagnostic accuracy but also informs tailored treatment strategies.
Genetic testing and family history analysis remain critical tools in managing MODY, offering patients like Emily a path to effective treatment and improved quality of life.
By addressing the question of how HNF1A mutations lead to MODY, this article provides a comprehensive overview for medical professionals and individuals seeking to understand this unique form of diabetes.
References: