How KCNJ11 Mutations Impair ATP-Sensitive Potassium Channel Function?
- admin
- December 25, 2024
- 5:53 pm
- No Comments
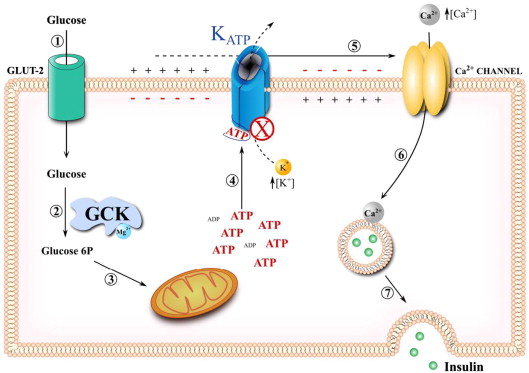
ATP-sensitive potassium (KATP) channels play a critical role in linking cellular energy status to membrane potential and insulin secretion, particularly in pancreatic beta cells.
Mutations in the KCNJ11 gene, which encodes the Kir6.2 subunit of the KATP channel, disrupt this essential function, leading to various metabolic disorders, including neonatal diabetes.
This article explores how KCNJ11 mutations impair KATP channel function, focusing on molecular mechanisms, physiological consequences, and real-life implications.
We will discuss the structure and role of the KATP channel, the effects of mutations on channel activity, and the broader impacts of these disruptions.
Article Index:
- Introduction to the KATP Channel and KCNJ11
- Structure and Function of the KATP Channel
- Molecular Mechanisms of KCNJ11 Mutations
- Types of KCNJ11 Mutations and Their Effects
- Physiological Implications of Impaired KATP Channel Function
- Real-Life Examples: Clinical Manifestations of KCNJ11 Mutations
- Scientific Evidence Supporting the Link Between KCNJ11 Mutations and KATP Channel Dysfunction
- Conclusion: The Impact of KCNJ11 Mutations on Cellular and Systemic Function
Introduction to the KATP Channel and KCNJ11
The KATP channel is a vital component of the cellular machinery that links metabolic activity to electrical signaling.
Located in pancreatic beta cells, it plays a critical role in regulating insulin secretion in response to blood glucose levels.
The channel comprises two subunits: Kir6.2, encoded by the KCNJ11 gene, and SUR1, encoded by the ABCC8 gene. Kir6.2 forms the potassium-conducting pore, while SUR1 modulates the channel’s activity by responding to metabolic signals, particularly changes in ATP and ADP levels.
Mutations in the KCNJ11 gene can impair the KATP channel’s ability to close in response to increased intracellular ATP levels, a mechanism essential for insulin secretion.
Normally, elevated ATP levels signal the channel to close, leading to membrane depolarization and the activation of voltage-dependent calcium channels. This allows calcium influx, which triggers insulin granule exocytosis.
However, KCNJ11 mutations, such as R201H and E23K, reduce ATP sensitivity, keeping the channel persistently open. This failure prevents membrane depolarization, disrupts calcium influx, and ultimately inhibits insulin release.
Scientific studies, including Gloyn et al. (2004) published in The New England Journal of Medicine, demonstrate how these mutations lead to conditions such as neonatal diabetes and developmental delay, epilepsy, and neonatal diabetes (DEND) syndrome.
Understanding these mechanisms is crucial for developing targeted therapies, such as sulfonylureas, which can restore insulin secretion by closing the mutant channels.
Structure and Function of the KATP Channel
The KATP channel acts as a metabolic sensor that plays a critical role in linking cellular energy levels to insulin secretion.
Under low-glucose conditions, the channel remains open, allowing potassium ions to flow out of the cell. This potassium efflux helps maintain the cell’s resting membrane potential, ensuring the cell is ready to respond to metabolic changes.
When glucose levels rise, the resulting increase in ATP production inhibits the KATP channel, causing it to close. This closure leads to membrane depolarization, which activates voltage-dependent calcium channels.
The subsequent calcium influx triggers the exocytosis of insulin granules, releasing insulin into the bloodstream to regulate blood glucose levels.
The Kir6.2 subunit, encoded by the KCNJ11 gene, plays a pivotal role in determining the channel’s sensitivity to ATP.
Mutations in KCNJ11 impair this finely tuned process by reducing ATP sensitivity or altering the channel’s ability to close.
This disruption leads to impaired insulin secretion and contributes to metabolic disorders, such as neonatal diabetes and DEND syndrome.
Studies like Proks et al. (2006) in Diabetes highlight the impact of these mutations on KATP channel functionality, underscoring their significant physiological consequences.
Molecular Mechanisms of KCNJ11 Mutations
Mutations in the KCNJ11 gene frequently result in a gain-of-function effect, rendering the ATP-sensitive potassium (KATP) channel less responsive to ATP.
This resistance to ATP prevents the channel from closing, even in the presence of high glucose levels that normally signal for closure.
As a result, the channel remains persistently open, leading to continuous potassium efflux.
This efflux maintains the pancreatic beta cell in a hyperpolarized state, effectively blocking the membrane depolarization required for calcium influx and insulin release.
Key Mechanisms:
- Altered ATP Binding Affinity: Mutations reduce the ability of ATP to bind effectively to the Kir6.2 subunit, impairing the channel’s responsiveness to metabolic changes.
- Enhanced Stability of the Open State: Certain mutations increase the likelihood of the channel remaining in its open conformation, further inhibiting proper closure.
- Disruption of Kir6.2-SUR1 Interaction: Mutations can impair the regulatory relationship between Kir6.2 and the SUR1 subunit, compromising the channel’s ability to modulate its activity.
These molecular alterations underscore how even single amino acid substitutions, such as those seen in mutations like R201H and V59M, can have profound consequences.
Research by Proks et al. (2006) in Diabetes highlights the extent to which these mutations disrupt KATP channel function, leading to conditions such as neonatal diabetes and DEND syndrome.
Types of KCNJ11 Mutations and Their Effects
Several KCNJ11 mutations have been identified, each exhibiting distinct effects on KATP channel function and corresponding clinical outcomes.
These mutations alter the channel’s ability to respond to ATP, leading to varying degrees of dysfunction and disease manifestations.
- E23K Mutation: This mutation increases channel activity by reducing its sensitivity to ATP. As a result, the channel remains open for longer durations, leading to persistent potassium efflux, membrane hyperpolarization, and hyperglycemia.
- R201H and R201C Mutations: These mutations are strongly associated with neonatal diabetes. They significantly impair ATP-induced channel closure, preventing the necessary depolarization for insulin secretion, resulting in chronic hyperglycemia.
- V59M Mutation: This severe mutation disrupts channel function more extensively, leading to conditions such as developmental delay, epilepsy, and neonatal diabetes (DEND syndrome). It affects not only pancreatic beta cells but also neurons, contributing to neurological symptoms.
Each mutation impacts the ATP-binding site or regulatory domains of the KATP channel, creating a spectrum of clinical phenotypes.
Studies, such as those by Gloyn et al. (2004), emphasize the relationship between these mutations and their varying impacts on channel function, highlighting their role in diverse metabolic and neurological disorders.
Physiological Implications of Impaired KATP Channel Function
The physiological consequences of KCNJ11 mutations are most prominently observed in pancreatic beta cells, where they disrupt normal insulin secretion, but their effects can also extend to other tissues, causing widespread complications.
- In Pancreatic Beta Cells: Mutations impair the function of ATP-sensitive potassium (KATP) channels, preventing proper membrane depolarization and calcium influx. This results in insufficient insulin secretion, leading to chronic hyperglycemia, a hallmark of neonatal diabetes.
- In the Brain: KCNJ11 mutations also affect KATP channels in neurons, contributing to neurological symptoms observed in conditions like developmental delay, epilepsy, and neonatal diabetes (DEND syndrome). Dysfunctional neuronal KATP channels disrupt normal brain activity, exacerbating these symptoms.
- Systemic Impact: Persistent hyperglycemia due to impaired insulin secretion can lead to long-term complications such as cardiovascular disease, diabetic nephropathy, and retinopathy. These secondary effects significantly impact the overall health and quality of life of affected individuals.
Understanding the multifaceted impact of KCNJ11 mutations is essential for developing effective therapeutic strategies to manage both metabolic and neurological outcomes, as highlighted in studies like Gloyn et al. (2004).
Clinical Manifestations of KCNJ11 Mutations
Let us walk you through a few such cases:
Case Study 1: Neonatal Diabetes in Infancy
Meet Maria, a bubbly two-month-old infant who presented with a rather serious health issue—severe hyperglycemia and dehydration.
Her parents were understandably worried as doctors ran tests to uncover the root cause. Genetic testing pinpointed the culprit: an R201H mutation in the KCNJ11 gene, confirming neonatal diabetes.
This mutation kept Maria’s KATP channels stubbornly open, preventing her beta cells from releasing insulin.
The good news?
Maria’s treatment team decided to transition her from insulin injections to sulfonylureas, a type of medication that coaxed the mutated channels to close.
The result was a remarkable recovery as her insulin secretion normalized, allowing her to thrive like any other infant.
Case Study 2: DEND Syndrome in a Young Child
Then there’s James, a curious and energetic five-year-old who faced more than just hyperglycemia.
Along with developmental delays and seizures, he was diagnosed with DEND syndrome, courtesy of a V59M mutation in his KCNJ11 gene.
This mutation affected not only the KATP channels in his pancreatic beta cells but also those in his neurons.
Sulfonylurea therapy came to the rescue, significantly improving his glycemic control.
However, managing James’ neurological symptoms required a multi-pronged approach, including physical therapy and specialized educational support.
While challenges remain, James’ story highlights the importance of understanding the broad impacts of KCNJ11 mutations.
Scientific Evidence Supporting the Link Between KCNJ11 Mutations and KATP Channel Dysfunction
Research has extensively demonstrated the impact of KCNJ11 mutations on KATP channel function:
- Gloyn et al., 2004: Identified KCNJ11 mutations as a cause of permanent neonatal diabetes and described their molecular mechanisms (The New England Journal of Medicine).
- Proks et al., 2006: Demonstrated how specific mutations alter ATP sensitivity, leading to impaired channel closure (Diabetes).
- Slingerland et al., 2006: Highlighted the phenotypic spectrum of KCNJ11 mutations, from isolated diabetes to syndromic presentations (Diabetologia).
These studies underscore the critical role of KCNJ11 in maintaining KATP channel function and its broader implications for health.
The Impact of KCNJ11 Mutations on Cellular and Systemic Function
Mutations in the KCNJ11 gene profoundly disrupt the function of ATP-sensitive potassium (KATP) channels, which act as critical gatekeepers linking cellular metabolism to electrical signaling in pancreatic beta cells.
These channels, responsible for regulating insulin secretion, become impaired due to mutations, leading to a cascade of metabolic dysfunctions.
The result is chronic hyperglycemia, a hallmark of neonatal diabetes, and in severe cases, developmental delay, epilepsy, and neonatal diabetes (DEND) syndrome. This disruption prevents the precise balance of potassium ion flow, keeping cells in a hyperpolarized state and effectively blocking insulin release.
Understanding the molecular mechanisms—such as impaired ATP binding and disrupted channel regulation—is essential for unraveling the complexity of these mutations.
Breakthroughs in genetic testing have significantly improved diagnostic accuracy, enabling earlier intervention.
Treatments like sulfonylureas have shown remarkable success in restoring channel function, offering renewed hope for affected families.
However, ongoing research strives to fine-tune therapies, addressing both metabolic and neurological symptoms to improve quality of life for those impacted by these challenging genetic disorders.
This effort underscores the profound importance of continued scientific exploration in translating genetic insights into lifesaving treatments.
References: