Insulin secretion is a finely tuned biological process critical for maintaining glucose homeostasis.
The potassium inward rectifier 6.2 (Kir6.2) channel, encoded by the KCNJ11 gene, plays a pivotal role in this mechanism as part of the ATP-sensitive potassium (KATP) channel in pancreatic beta cells.
Mutations in the Kir6.2 protein disrupt the channel’s function, impairing insulin release and leading to conditions like neonatal diabetes or permanent neonatal diabetes mellitus (PNDM).
In this article, we explore how and why mutations in Kir6.2 lead to defective insulin secretion, providing real-life examples and supporting scientific research.
In This Article:
- Introduction to Kir6.2 and Its Role in Insulin Secretion
- Mechanism of KATP Channels in Beta Cells
- Impact of Kir6.2 Mutations on Channel Function
- 3.1. Loss of ATP Sensitivity
- 3.2. Persistent Channel Opening
- Beta-Cell Dysfunction Due to Kir6.2 Mutations
- 4.1. Impact on Membrane Depolarization
- 4.2. Impaired Calcium Influx and Insulin Granule Exocytosis
- Case Studies of Neonatal Diabetes from Kir6.2 Mutations
- 5.1. Case Study: A Newborn with R201H Mutation
- 5.2. Case Study: Family with Inherited KCNJ11 Mutation
- Scientific Studies Supporting Kir6.2 Mutations in Insulin Dysregulation
- FAQs on Neonatal Diabetes from Kir6.2 Mutations
Introduction to Kir6.2 and Its Role in Insulin Secretion
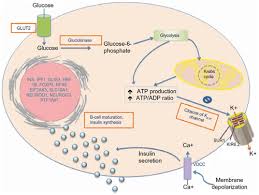
Kir6.2, encoded by the KCNJ11 gene, is a key subunit of the ATP-sensitive potassium (KATP) channel in pancreatic beta cells.
This channel works in tandem with the sulfonylurea receptor 1 (SUR1), encoded by the ABCC8 gene, to regulate beta-cell membrane potential.
The Kir6.2-SUR1 complex functions as a metabolic sensor, coupling intracellular ATP and ADP levels to insulin secretion.
When ATP levels rise due to glucose metabolism, the KATP channel closes, triggering membrane depolarization and insulin release.
Research published in Nature Genetics (Gloyn et al., 2004) underscores the critical role of Kir6.2 in maintaining glucose homeostasis.
Mechanism of KATP Channels in Beta Cells
The KATP channel plays a pivotal role in linking glucose metabolism to insulin secretion.
In response to rising blood glucose levels, pancreatic beta cells metabolize glucose to generate ATP.
Elevated ATP concentrations close the KATP channels, halting the efflux of potassium ions.
This closure depolarizes the cell membrane, which then activates voltage-gated calcium channels, allowing calcium ions to flow into the cell.
Calcium influx acts as a signal for insulin granules to undergo exocytosis, releasing insulin into the bloodstream to regulate glucose levels.
Kir6.2 mutations, such as those found in the KCNJ11 gene, disrupt this delicately balanced mechanism.
These mutations impair the sensitivity of KATP channels to ATP, keeping them persistently open.
As a result, membrane depolarization does not occur, preventing calcium influx and stalling insulin secretion. Conversely, some mutations may cause excessive channel closure, leading to dysregulated insulin release.
A study in Endocrine Reviews (Ashcroft, 2006) highlights these disruptions as critical contributors to neonatal diabetes or hypoglycemic disorders.
Impact of Kir6.2 Mutations on Channel Function
A quick look at this unique process:
Loss of ATP Sensitivity:
Kir6.2 mutations often impair the ATP-sensing ability of the KATP channel.
Normally, ATP binds to Kir6.2, closing the channel and enabling insulin secretion. Mutations such as R201H reduce ATP binding, leaving the channel persistently open.
As a result, beta cells fail to depolarize, preventing the calcium influx needed for insulin release.
A study published in Nature Genetics (Gloyn et al., 2004) demonstrated that reduced ATP sensitivity is a hallmark of neonatal diabetes caused by Kir6.2 mutations.
Persistent Channel Opening:
Kir6.2 mutations can also increase the channel’s intrinsic open probability.
For example, the V59M mutation enhances the channel’s responsiveness to magnesium-bound ADP, counteracting ATP-mediated closure.
Persistent channel opening keeps the beta cells hyperpolarized, rendering them unresponsive to glucose.
Beta-Cell Dysfunction Due to Kir6.2 Mutations
Here is how it works:
Disruption of Membrane Depolarization: A Core Mechanism:
Kir6.2 mutations in the KCNJ11 gene fundamentally disrupt membrane depolarization in beta cells.
Under normal physiological conditions, glucose metabolism generates ATP, which binds to and closes KATP channels.
This closure leads to membrane depolarization, activating voltage-gated calcium channels and triggering calcium influx.
However, Kir6.2 mutations, such as R201H and V59M, impair the sensitivity of KATP channels to ATP, causing them to remain open despite high intracellular ATP levels.
This persistent activity maintains the beta cell membrane in a hyperpolarized state, preventing calcium channels from opening.
Without calcium entry, the signaling cascade for insulin granule exocytosis is interrupted, halting insulin secretion.
A study in Nature Genetics (Gloyn et al., 2004) emphasized this mechanism as the cornerstone of insulin secretion failure in neonatal diabetes.
Calcium Influx and Insulin Granule Exocytosis: The Missing Link
Calcium influx is the trigger for insulin granule exocytosis, where insulin is released into the bloodstream.
Kir6.2 mutations block this critical step by impairing calcium signaling.
Persistent KATP channel activity leads to insufficient intracellular calcium levels, creating a cascade of dysfunction.
This directly translates to hyperglycemia and glucose intolerance, the primary features of neonatal diabetes.
Research published in Diabetes (Ashcroft & Rorsman, 2012) detailed how mutations in Kir6.2 disrupt the calcium-insulin pathway, offering a foundation for understanding the pathophysiology of the condition.
This understanding has paved the way for treatments like sulfonylureas, which pharmacologically close KATP channels to restore beta-cell function.
Case Studies of Neonatal Diabetes from Kir6.2 Mutations
A quick look at 2 case studies that shall keep you updated with this topic:
A Newborn’s Struggle with R201H Mutation: Early Diagnosis and Intervention:
A newborn boy exhibited severe hyperglycemia just days after birth, raising concerns of neonatal diabetes.
Genetic analysis pinpointed the R201H mutation in the KCNJ11 gene, a defect known to impair ATP sensitivity and keep the KATP channels persistently open.
This disruption prevented beta-cell depolarization and calcium influx, halting insulin secretion entirely.
The infant was transitioned from insulin injections to sulfonylureas, a medication class that pharmacologically closes KATP channels.
Within weeks, the treatment restored his insulin secretion and stabilized blood glucose levels.
This case illustrates the life-changing potential of early genetic testing in identifying Kir6.2-related diabetes and tailoring precise interventions.
A study published in Diabetologia (Hattersley et al., 2006) demonstrated that such targeted therapy is not only effective but also crucial for improving long-term outcomes.
Genetic Inheritance: A Family’s Experience with the V59M Mutation:
In a family spanning three generations, several members showed varying degrees of diabetes onset.
Genetic testing revealed the V59M mutation in the KCNJ11 gene in all affected individuals.
For some, the mutation caused neonatal diabetes, while others experienced a milder, late-onset form resembling MODY (maturity-onset diabetes of the young).
This variability underscores the interplay between genetic predisposition and environmental influences.
While neonates presented with insulin dependency, older family members managed their condition with lifestyle modifications and oral medications.
Research in the Journal of Physiology (Tammaro et al., 2008) highlights how such mutations alter KATP channel gating, resulting in varied clinical manifestations.
These cases emphasize the necessity of genetic counseling and tailored treatment plans for families with inherited KCNJ11 mutations.
Scientific Studies Supporting Kir6.2 Mutations in Insulin Dysregulation
- Gloyn et al., 2004 (Nature Genetics):
This foundational research identified KCNJ11 mutations, such as R201H, as a significant cause of permanent neonatal diabetes mellitus (PNDM). The study highlighted the molecular mechanism behind ATP-insulin coupling defects caused by these mutations, underscoring their critical role in impaired insulin secretion. - Ashcroft & Rorsman, 2012 (Diabetes):
This study delved into how mutations in 2 affect KATP channel function in beta cells. It revealed that persistent channel activity, due to impaired ATP sensitivity, disrupts calcium signaling, a key step in insulin granule exocytosis. - Hattersley et al., 2006 (New England Journal of Medicine):
Exploring therapeutic avenues, this research demonstrated the efficacy of sulfonylureas in closing mutant KATP channels. The findings provided a personalized treatment framework for patients with Kir6.2-related neonatal diabetes. - Tammaro et al., 2008 (Journal of Physiology):
Focusing on the gating properties of KATP channels, this study showed how mutations like V59M increase the channel’s open probability, preventing proper beta-cell depolarization. This disruption hinders the critical calcium influx necessary for insulin release.
Together, these studies form a robust body of evidence linking Kir6.2 mutations to disrupted insulin secretion, paving the way for targeted therapies and better clinical outcomes.
FAQs on Neonatal Diabetes from Kir6.2 Mutations
Q-1: What role does Kir6.2 normally play in insulin secretion—and what breaks when it mutates?
A-1: Kir6.2 forms the pore of the KATP channel in β-cells. When glucose rises, ATP increases, KATP closes, the membrane depolarizes, calcium enters, and insulin is released. Mutations that reduce ATP sensitivity keep KATP too open, blocking depolarization and calcium entry—so insulin release under normal glucose is blunted or absent.
Q-2: Do all Kir6.2 mutations reduce insulin, or can some cause too much insulin?
A-2: Both happen. Most gain-of-function variants (ATP-insensitive) cause under-secretion and diabetes, often from infancy. Some loss-of-function variants (excessive closure) drive unregulated depolarization and hyperinsulinism with hypoglycemia. The clinical picture—diabetes vs. hypoglycemia—often maps to whether KATP is pathologically open or closed at physiologic ATP.
Q-3: How do Kir6.2 mutations alter the channel’s “coaching staff” (SUR1, Mg-ADP, PIP₂) to derail glucose sensing?
A-3: Kir6.2 gating is tuned by SUR1, Mg-ADP (which favors opening), and PIP₂ at the membrane. Mutations can strengthen open-state stabilization (PIP₂/Mg-ADP bias) or weaken ATP inhibition, shifting the channel toward persistent openness. Others impair subunit coupling or trafficking, reducing correctly assembled channels and distorting the normal ATP-to-insulin translation.
Q-4: Why do some Kir6.2 mutations also affect the brain and muscles, not just β-cells?
A-4: The same KATP architecture exists in neurons and muscle. Variants that strongly favor the open state can dampen neuronal excitability or alter motor unit function, leading to features like developmental delay, seizures, or hypotonia alongside disordered insulin secretion—because the gating error is shared across tissues.
Q-5: How do these channel defects reshape the pattern of insulin release across a meal?
A-5: With KATP stuck too open, β-cells fail to generate the rapid depolarization needed for first-phase insulin, so the early “spike” after eating is lost. Second-phase, which depends on sustained calcium oscillations and granule trafficking, is also reduced. The result is higher post-meal glucose excursions despite adequate β-cell mass.
Q-6: What makes sulfonylureas, diazoxide, or GLP-1 agents succeed or fail with Kir6.2 mutations?
A-6: Sulfonylureas bind SUR1 to close KATP despite ATP insensitivity—effective when channels reach the surface and coupling remains intact. Diazoxide does the opposite (opens KATP), useful in hyperinsulinism due to over-closure. GLP-1–based therapies can boost residual secretion downstream, but if KATP cannot close at all, their effect is limited. Drug response often tracks the specific biophysical change the mutation causes.
Conclusion
Kir6.2 mutations disrupt insulin secretion by impairing ATP sensitivity, causing persistent KATP channel activity, and preventing beta-cell depolarization.
As per bestdietarysupplementfordiabetics.com, “These mutations lead to hyperglycemia and neonatal diabetes, conditions that can significantly impact early development and long-term health”.
By understanding the molecular mechanisms underlying these mutations, clinicians and researchers can develop targeted treatments, such as sulfonylureas, to restore beta-cell function.
Continued research is essential for unraveling the complex genetic and physiological factors contributing to Kir6.2-related disorders, paving the way for improved therapeutic strategies.
References:
admin
All Posts