How SUR1 Mutations Impair Insulin Secretion?
- admin
- December 19, 2024
- 6:29 am
- No Comments
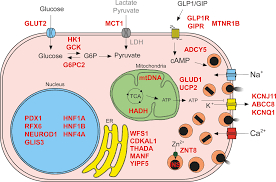
The proper functioning of insulin secretion in pancreatic beta cells is crucial for glucose homeostasis.
The sulfonylurea receptor 1 (SUR1), encoded by the ABCC8 gene, is a vital subunit of the ATP-sensitive potassium (KATP) channel in beta cells.
Mutations in SUR1 can severely impair insulin secretion, leading to conditions like neonatal diabetes or congenital hyperinsulinism.
This article by bestdietarysupplementfordiabetics.com delves into how SUR1 mutations disrupt insulin secretion, exploring the underlying mechanisms, the clinical implications, and real-life case studies.
Article Index:
- Role of SUR1 in Insulin Secretion
- Mechanisms of SUR1 Mutations Affecting Beta Cells
- 2.1. Dysfunctional KATP Channels
- 2.2. Altered ATP Sensitivity
- 2.3. Impaired Glucose Responsiveness
- Clinical Implications of SUR1 Mutations
- 3.1. Neonatal Diabetes
- 3.2. Congenital Hyperinsulinism
- Case Studies
- 4.1. A Newborn with a SUR1 Mutation
- 4.2. Familial SUR1 Mutation and Variable Manifestations
- FAQs on SUR1 & Insulin Secretion
- Conclusion
Role of SUR1 in Insulin Secretion
The sulfonylurea receptor 1 (SUR1), encoded by the ABCC8 gene, is a vital subunit of the ATP-sensitive potassium (KATP) channel in pancreatic beta cells.
It partners with Kir6.2, encoded by the KCNJ11 gene, to form a channel that regulates the membrane potential by controlling potassium ion flux.
This complex acts as a metabolic sensor, ensuring insulin is secreted in response to rising glucose levels.
Under normal conditions, an increase in blood glucose results in ATP production. ATP binds to the KATP channel, causing it to close.
This closure depolarizes the beta cell membrane, activating voltage-gated calcium channels. Calcium influx triggers the exocytosis of insulin granules, effectively regulating blood glucose.
Mutations in SUR1 impair this mechanism, either by preventing KATP channel closure or disrupting ATP binding. This leads to conditions like neonatal diabetes or congenital hyperinsulinism, depending on the nature of the mutation.
Scientific studies support this mechanism. Ashcroft & Rorsman (2002) in Nature Genetics highlighted the role of KATP channels in beta-cell physiology, while Tammaro et al. (2008) in Diabetes demonstrated how SUR1 mutations alter ATP sensitivity, impairing insulin secretion.
Understanding this finely tuned mechanism is critical for developing targeted therapies.
Mechanisms of SUR1 Mutations Affecting Beta Cells
A quick look at the process:
Dysfunctional KATP Channels:
SUR1 mutations often result in defective KATP channel function.
A study in Nature Genetics (Ashcroft & Rorsman, 2002) highlighted that loss-of-function mutations render the channel incapable of opening, trapping the beta cell in a depolarized state.
This causes inappropriate insulin secretion, even during low glucose levels, as seen in congenital hyperinsulinism.
Conversely, gain-of-function mutations keep the KATP channel persistently open, preventing membrane depolarization. This inhibits insulin release despite high blood glucose, leading to neonatal diabetes.
Altered ATP Sensitivity:
ATP sensitivity is critical for KATP channel closure.
Mutations in SUR1 often alter the channel’s responsiveness to ATP, making it insensitive to physiological glucose changes.
Research in Diabetes (Tammaro et al., 2008) demonstrated that gain-of-function SUR1 mutations impair ATP binding, leading to chronic channel activity and reduced insulin secretion.
Impaired Glucose Responsiveness:
SUR1 mutations also disrupt the beta cell’s ability to respond to fluctuating glucose levels.
A study published in The Journal of Clinical Investigation (Hattersley et al., 2006) showed that mutations impair the channel’s ability to transition between open and closed states.
This lack of dynamic response leads to blunted insulin secretion even during hyperglycemia.
Clinical Implications of SUR1 Mutations
Let us walk you through the clinical process:
Neonatal Diabetes: A Role for Gain-of-Function Mutations:
Neonatal diabetes is a rare but severe condition diagnosed within the first six months of life.
It arises from genetic mutations that impair insulin secretion. Gain-of-function mutations in the SUR1 protein are a key contributor to this condition.
These mutations lead to KATP channels that remain persistently open, even in the presence of high ATP levels, preventing beta-cell membrane depolarization and calcium influx required for insulin secretion.
The landmark study by Gloyn et al. (2004) in Nature Genetics established SUR1 mutations as a major cause of both transient and permanent neonatal diabetes.
This research emphasized the importance of genetic testing for early diagnosis and personalized treatment, such as transitioning from insulin injections to sulfonylureas in cases with ATP-sensitive KATP channel dysfunction.
The findings underscore how dysfunctional KATP channels disrupt glucose homeostasis from infancy.
Congenital Hyperinsulinism: Loss-of-Function Consequences:
In contrast, loss-of-function mutations in SUR1 result in congenital hyperinsulinism (CHI), where the KATP channels remain closed, even under low glucose conditions.
This causes unregulated insulin release, leading to persistent hypoglycemia—a condition that can be life-threatening if not managed promptly.
Research by Stanley et al. (2016) in Pediatric Diabetes demonstrated that severe CHI cases often require surgical intervention, such as partial pancreatectomy, when medical therapies fail to control hypoglycemia.
These studies highlight how distinct mutations in SUR1 can cause opposing metabolic disorders, each requiring tailored interventions.
Liam’s Journey: A Newborn with a SUR1 Mutation
At just three months old, Liam was diagnosed with severe hyperglycemia, a rare but alarming condition in neonates.
Initial attempts to manage his blood sugar with traditional insulin therapies proved challenging.
Genetic testing revealed a gain-of-function mutation in the ABCC8 gene, which encodes the SUR1 protein.
This mutation disrupted the delicate ATP-sensitive KATP channel function in pancreatic beta cells, keeping the channels perpetually open.
As a result, Liam’s beta cells failed to depolarize, effectively halting calcium influx and insulin secretion.
Liam’s endocrinologist prescribed sulfonylureas, a class of drugs designed to close KATP channels, bypassing the ATP-binding defect caused by the mutation.
Remarkably, within weeks of starting the treatment, Liam’s glucose levels stabilized, and his insulin secretion partially recovered.
His case highlights how targeted genetic therapies can transform the prognosis for neonatal diabetes patients, bridging the gap between genetic diagnosis and personalized treatment.
The Complexity of Familial SUR1 Mutations:
The story of the O’Connell family adds another layer of intrigue to SUR1 mutations.
Genetic testing revealed that both the father, Richard, and his daughter, Ella, carried a shared SUR1 mutation. However, the clinical outcomes were strikingly different.
Richard developed early-onset diabetes, requiring lifelong insulin therapy, while Ella exhibited only mild glucose intolerance, managed effectively with dietary modifications.
This variability in clinical presentation underscores the interplay of genetic modifiers and environmental factors.
For the O’Connells, their story emphasizes the nuanced impact of SUR1 mutations and the importance of personalized care tailored to individual manifestations.
FAQs on SUR1 & Insulin Secretion
Q-1: Mechanistically, how do SUR1 (ABCC8) mutations blunt glucose-stimulated insulin secretion?
A-1: SUR1 is the regulatory subunit of the pancreatic β-cell KATP channel. Normally, rising ATP from glucose metabolism closes KATP, the membrane depolarizes, calcium rushes in, and insulin is released. Many SUR1 mutations skew this sequence by making channels too easy to keep open (through altered nucleotide/PIP₂ effects) or too hard to close with ATP. With KATP persistently open, depolarization stalls, calcium entry falls, and first-phase insulin release collapses.
Q-2: Why do some SUR1 variants cause diabetes while others cause congenital hyperinsulinism (CHI)?
A-2: It’s the direction of channel change. Gain-of-function variants keep KATP overly active, hyperpolarizing β-cells and suppressing insulin—producing neonatal/monogenic diabetes. Loss-of-function variants prevent KATP from opening, driving unchecked depolarization and calcium influx—producing CHI and hypoglycemia. The same gene thus sits at the crossroads of too little versus too much insulin secretion depending on how KATP gating is altered.
Q-3: How do SUR1 mutations modify responses to drugs like sulfonylureas or diazoxide?
A-3: Sulfonylureas bind SUR1 and favor KATP closure. Many diabetes-causing SUR1 variants remain sulfonylurea-responsive, allowing patients to replace or reduce insulin injections with oral therapy. Diazoxide does the opposite—stabilizes KATP in the open state—to treat hyperinsulinism. Severe loss-of-function SUR1 mutations can make CHI diazoxide-unresponsive, necessitating alternatives (e.g., octreotide, surgery). Drug response often mirrors the specific gating defect.
Q-4: Do SUR1 mutations ever reduce channel numbers at the cell surface instead of just changing gating?
A-4: Yes. Some variants disrupt folding, assembly, or trafficking of the SUR1–Kir6.2 complex, so fewer channels reach the membrane. With reduced surface expression, the β-cell’s excitability set-point shifts even if the remaining channels gate normally. This “less hardware” mechanism—alongside altered gating—can lower insulin secretion by limiting how effectively metabolism controls electrical activity.
Q-5: What clinical clues suggest a SUR1 mutation is behind insulin problems, and how can structure guide interpretation?
A-5: Clues include antibody-negative hyperglycemia presenting in infancy or childhood, low birth weight, a family history of neonatal diabetes or hyperinsulinism, and striking sensitivity (or resistance) to sulfonylureas/diazoxide that seems “too good” (or too poor) to be coincidental. Knowledge of SUR1’s architecture—its nucleotide-binding domains, transmembrane regions, and drug-binding pockets—helps predict whether a given variant will keep KATP too open, too closed, or off the cell surface, which in turn informs therapy choice and prognosis.
Bottom line: SUR1 mutations impair insulin secretion by skewing KATP gating, breaking nucleotide signaling, or reducing channel trafficking. Depending on direction, the same protein can cause diabetes or hyperinsulinism—and the pattern of drug responsiveness often reveals the underlying defect.
Takeaway:
The complex interplay between SUR1 mutations and insulin secretion underscores the pivotal role of KATP channel function in maintaining glucose homeostasis.
These mutations impair ATP sensitivity, hinder glucose responsiveness, and disrupt membrane depolarization, resulting in diverse conditions like neonatal diabetes and congenital hyperinsulinism.
Gain-of-function mutations keep KATP channels persistently open, inhibiting insulin release, while loss-of-function mutations cause unregulated insulin secretion.
Early genetic diagnosis enables precise interventions, such as sulfonylurea therapy, which can restore insulin secretion by bypassing the molecular defect.
Research, including Gloyn et al. (2004) and Stanley et al. (2016), highlights the transformative potential of these targeted approaches for improved patient outcomes.
References: