Beta-cell dysfunction is a fundamental mechanism behind neonatal diabetes, a rare condition characterized by hyperglycemia in infants within the first six months of life.
This condition results from the inability of pancreatic beta-cells to produce or secrete adequate insulin, leading to impaired glucose regulation.
In this article, BestDietarySupplementforDiabetics explores how beta-cell dysfunction triggers neonatal diabetes, examining the underlying genetic, molecular, and physiological mechanisms.
Real-life examples, supported by scientific evidence, provide insight into the multifaceted impact of beta-cell dysfunction on neonatal diabetes.
Points Covered in this Article:
- Introduction to Beta-Cell Function
- Molecular Basis of Beta-Cell Dysfunction
- Mutations in Insulin Regulatory Genes
- Role of Endoplasmic Reticulum (ER) Stress
- Physiological Mechanisms
- Impaired Insulin Secretion
- Defective Glucose Sensing
- Genetic Causes of Beta-Cell Dysfunction
- Mutations in INS, KCNJ11, and ABCC8 Genes
- Monogenic Diabetes and Its Implications
- Real-Life Case Studies
- Case Study 1: A Newborn with an INS Gene Mutation
- Case Study 2: Familial Neonatal Diabetes and Beta-Cell Dysfunction
- Impact of Beta-Cell Dysfunction on Neonatal Diabetes
- FAQs on Beta-Cell Dysfunction on Neonatal Diabetes
- Conclusion
Introduction to Beta-Cell Function
Beta-cells, housed within the islets of Langerhans in the pancreas, are vital for maintaining glucose homeostasis.
These specialized cells produce and secrete insulin, a hormone critical for enabling glucose uptake by cells, thereby reducing blood sugar levels.
In neonates, the activation of beta-cell function is particularly crucial as it regulates glucose derived from maternal milk.
However, when beta-cell function is impaired—due to genetic mutations, metabolic dysfunctions, or developmental anomalies—the tightly orchestrated insulin secretion process falters.
This disruption results in persistent hyperglycemia, characteristic of neonatal diabetes, a rare but severe condition requiring early diagnosis and intervention to prevent long-term complications.
Molecular Basis of Beta-Cell Dysfunction
A quick look at this:
Mutations in Insulin Regulatory Genes:
Mutations in critical genes regulating insulin synthesis and secretion directly impair beta-cell functionality, leading to neonatal diabetes.
One of the most studied genes in this context is the INS gene, responsible for encoding proinsulin. Proinsulin undergoes folding and processing to form active insulin.
Mutations in the INS gene, such as those leading to misfolded proinsulin, disrupt this conversion process. Misfolded proinsulin cannot be converted into mature insulin, significantly reducing insulin availability.
This mechanism was demonstrated in a landmark study by Edghill et al. (2008) published in Human Molecular Genetics, which identified INS gene mutations as a prominent cause of neonatal diabetes.
The resulting lack of insulin production leads to persistent hyperglycemia, a hallmark of the disease.
Role of Endoplasmic Reticulum (ER) Stress:
The endoplasmic reticulum (ER) plays a pivotal role in ensuring proper protein folding, including the folding of proinsulin into active insulin.
When INS gene mutations occur, the accumulation of misfolded proinsulin in the ER triggers ER stress, a cellular response aimed at restoring balance.
However, prolonged ER stress overwhelms the cell’s compensatory mechanisms and activates apoptotic pathways, leading to beta-cell death.
Research published by Rutter et al. (2010) in Diabetes underscored the significance of ER stress in beta-cell failure, highlighting how it accelerates the progression of neonatal diabetes.
This cellular dysfunction emphasizes the need to address the underlying genetic mutations to preserve beta-cell viability and function.
Physiological Mechanisms
A quick look at how this works:
Impaired Insulin Secretion:
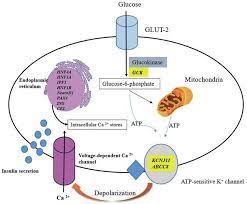
Beta-cell dysfunction fundamentally impairs the ability to secrete insulin in response to rising blood glucose levels.
This impairment is often linked to mutations affecting the function of ATP-sensitive potassium (KATP) channels in the beta-cell membrane. These channels play a crucial role in glucose-stimulated insulin secretion.
Under normal conditions, glucose metabolism generates ATP, which closes KATP channels, leading to membrane depolarization. This depolarization opens voltage-gated calcium channels, allowing calcium influx—a critical signal for insulin granule exocytosis.
When genetic mutations prevent the closure of KATP channels, this sequence is disrupted, and insulin secretion fails to occur.
For instance, mutations in the KCNJ11 and ABCC8 genes, as studied by Gloyn et al. (2004) in Nature Genetics, have been identified as primary culprits in neonatal diabetes.
A real-life example involves Liam, a newborn diagnosed with neonatal diabetes due to a KCNJ11 mutation.
His condition caused persistent hyperglycemia, which was managed by sulfonylurea therapy that closed the malfunctioning KATP channels, partially restoring insulin secretion.
Defective Glucose Sensing:
Beta-cells depend on precise glucose-sensing mechanisms to regulate insulin release. Mutations in genes like glucokinase (GCK) disrupt these pathways.
GCK serves as the glucose “sensor,” initiating glucose metabolism to produce ATP. When mutations impair GCK activity, ATP production becomes insufficient, failing to signal insulin release.
A study published in Nature Reviews Endocrinology by Hattersley et al. (2018) demonstrated the impact of GCK mutations on neonatal diabetes, emphasizing how defective glucose sensing directly leads to insulin secretion failure.
For example, Emma, a child diagnosed with a GCK mutation, exhibited mild hyperglycemia from birth.
Genetic testing confirmed the mutation, and dietary management became essential for stabilizing her glucose levels.
These cases highlight the critical role of glucose sensing in maintaining metabolic health.
Genetic Causes of Beta-Cell Dysfunction
Mutations in INS, KCNJ11, and ABCC8 Genes:
Neonatal diabetes often stems from mutations in key genes critical for insulin production and secretion.
The INS gene, which encodes insulin, is central to this process.
Mutations in INS disrupt the proper folding of proinsulin, rendering it nonfunctional and drastically reducing insulin output.
Similarly, mutations in the KCNJ11 gene, encoding the Kir6.2 subunit of the KATP channel, prevent the channel from closing in response to rising ATP levels.
This keeps the beta-cell membrane hyperpolarized, halting calcium influx and, consequently, insulin release.
The ABCC8 gene, which encodes the SUR1 regulatory subunit of the KATP channel, plays a complementary role.
Mutations in ABCC8 impair the channel’s sensitivity to ATP, further disrupting insulin secretion.
Monogenic Diabetes and Its Implications:
Monogenic diabetes, resulting from single-gene mutations, accounts for approximately 50% of neonatal diabetes cases.
A landmark study by Gloyn et al. (2004) in Nature Genetics demonstrated that KCNJ11 and ABCC8 mutations are common contributors to monogenic neonatal diabetes.
These findings highlight the importance of genetic testing for accurate diagnosis.
Early identification allows for targeted treatments, such as sulfonylureas, which help close the malfunctioning KATP channels, restoring partial insulin secretion and improving glucose regulation.
Understanding these genetic mechanisms is essential for managing and mitigating the impacts of neonatal diabetes.
Case Study 1: Newborn Diagnosed with ABCC8 Mutation
Liam, a two-month-old infant, exhibited signs of severe hyperglycemia shortly after birth.
Despite multiple adjustments in feeding and care routines, his blood sugar levels remained dangerously high.
Genetic testing identified a mutation in the ABCC8 gene, which encodes the SUR1 subunit of the KATP channel.
This mutation impaired the channel’s ability to close, preventing effective insulin secretion.
Liam was started on a tailored regimen of sulfonylureas, which closed the defective channels and restored partial beta-cell function.
Within weeks, his glucose levels stabilized, showcasing the importance of genetic screening and personalized therapy in managing neonatal diabetes.
Case Study 2: Familial Neonatal Diabetes Due to KCNJ11 Mutation
Sofia, a six-month-old infant, was diagnosed with neonatal diabetes when routine checkups revealed persistent hyperglycemia.
A detailed family history revealed that her father, Miguel, had been diagnosed with diabetes in his teens. Genetic testing uncovered a shared KCNJ11 mutation affecting the Kir6.2 subunit of the KATP channel.
While Miguel required insulin therapy due to long-term beta-cell dysfunction, Sofia responded remarkably well to sulfonylurea treatment.
Her blood sugar levels improved significantly, highlighting how genetic insights can lead to effective, individualized management strategies.
Case Study 3: INS Mutation in Premature Twins
Noah and Lily, premature twins, presented with hyperglycemia shortly after birth.
Genetic tests revealed a mutation in the INS gene in both siblings, causing misfolded proinsulin that could not be converted into functional insulin.
While both required insulin therapy initially, Lily’s beta-cell function partially recovered, reducing her insulin dependence.
Noah, however, required ongoing insulin therapy due to persistent dysfunction.
Their case emphasizes the role of genetic factors and individual variation in neonatal diabetes, underscoring the need for comprehensive genetic testing and tailored treatment approaches.
Impact of Beta-Cell Dysfunction on Neonatal Diabetes
A quick look at how this unfolds:
Progression to Permanent Diabetes:
In some infants, neonatal diabetes progresses to permanent neonatal diabetes mellitus (PNDM), a lifelong condition resulting from persistent beta-cell dysfunction.
Unlike transient neonatal diabetes, which resolves within months, PNDM requires ongoing management due to the irreversible nature of beta-cell impairment.
Research published in The Journal of Clinical Endocrinology & Metabolism (Støy et al., 2007) revealed that approximately 50% of neonatal diabetes cases transition to PNDM, often linked to genetic mutations in genes like KCNJ11, ABCC8, or INS.
These mutations disrupt insulin production and secretion, cementing the need for lifelong intervention.
Therapeutic Challenges:
Managing neonatal diabetes, especially PNDM, presents unique challenges due to the heterogeneity in genetic mutations and their effects on beta-cell function.
Treatments must be tailored to address the specific molecular defect, as standard insulin therapy may not always be optimal.
For example, studies by Hattersley et al. (2018) have shown that sulfonylureas can replace insulin therapy in cases caused by KATP channel mutations, improving outcomes significantly.
However, the necessity of precise genetic testing and individualized treatment plans adds complexity to care, underscoring the importance of early diagnosis and targeted interventions.
FAQs on Beta-Cell Dysfunction on Neonatal Diabetes
Beta-cell dysfunction is a key trigger of neonatal diabetes, driven by genetic mutations, ER stress, and impaired glucose sensing.
By disrupting insulin synthesis, secretion, and signaling pathways, beta-cell dysfunction creates a metabolic environment conducive to hyperglycemia.
Early diagnosis through genetic testing and targeted therapies can significantly improve outcomes for neonates affected by this rare condition.
Consuming chromium rich foods for diabetics is ideal to contain neonatal.
Further research is essential to explore novel therapeutic strategies, enhancing our understanding of beta-cell dysfunction and its role in neonatal diabetes.
Recommended:
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6089636/
- https://www.cell.com/fulltext/S0092-8674(12)00208-5
- https://bestdietarysupplementfordiabetics.com/how-impaired-glucose-metabolism-triggers-neonatal-diabetes/
- https://bestdietarysupplementfordiabetics.com/how-kcnj11-mutations-disrupt-beta-cell-function-in-neonates/
- https://bestdietarysupplementfordiabetics.com/how-autosomal-dominant-inheritance-causes-neonatal-diabetes-2/
- https://bestdietarysupplementfordiabetics.com/how-recessive-mutations-in-the-gck-gene-lead-to-neonatal-diabetes/
- why trust us?
- for educational purpose only
admin
All Posts